轨迹分析的软件有很多,其中最大名气的当属 Monocle2 和 Monocle3 ,但是他们的作图风格嘛,确实差点意思。后来我遇到了 slingshot 让我眼前一亮,今天就让我们来看看 slingshot 到底应该如何实现。
slingshot 的官方教程链接:
https://bioconductor.org/packages/release/bioc/vignettes/slingshot/inst/doc/vignette.html
今天我们就来实现下面这个图:
前言
最近跟群里的小伙伴交流的时候,发现有些初学者对于拟时序分析缺乏一些基本的概念。在这里,有必要简单讲讲为什么要做拟时序分析?
- 拟时序分析的基本假设:有机生物体内有成千上亿的细胞,他们扮演不同的身份、执行不同的功能。但我们都知道,他们并不是泾渭分明的,而是存在过渡态。比如骨髓干细胞(HSC)会逐步分化为髓系、淋巴系、红系等,我们称这样的分化轨迹为
lineage,这里面就会存在大量不同分化状态的细胞。 - 什么时候可以做拟时序: 当你的样本或研究对象在生物学上存在过渡态细胞时,就可以做拟时序分析。
- 拟时序分析能得到什么: 最重要的结果,就是得到拟合的“假时序”,我们可以根据合理的”假时许“对基因表达的变化值进行推断。此外,我们结合
CytoTRACE、palantir等算法评估细胞干性,可以找到细胞分化的”变异点“,并找到什么基因/基因集推动这样的改变,这一部分小编还在学习过程,未来有机会的话再分享。
今天我们使用的数据是 DISCO 图谱网站上的骨髓单细胞测序数据,该数据集包含了28万细胞,为了更方便地进行实操,我们挑选了以 HSC 为起点的髓系和红系数据,并随机挑选了15000个细胞,作为我们的示例数据。
示例数据
链接:https://pan.baidu.com/s/1pPp4pOnYbIwW30EFi8xbRg?pwd=joe1
提取码:joe1
里面有两个压缩包:
- 无原始数据(已经处理好的,可以不跑data_prepare这一步)
- 含原始数据(文件较大,处理过程保存在data_prepare,你可以按照原始数据再重新构建一个新的练习数据)
【建议】
你可以先下载无原始数据的,用我处理好的数据先跑下来,避免下载太久
代码实操
1. 构建SingleCellExperiment对象
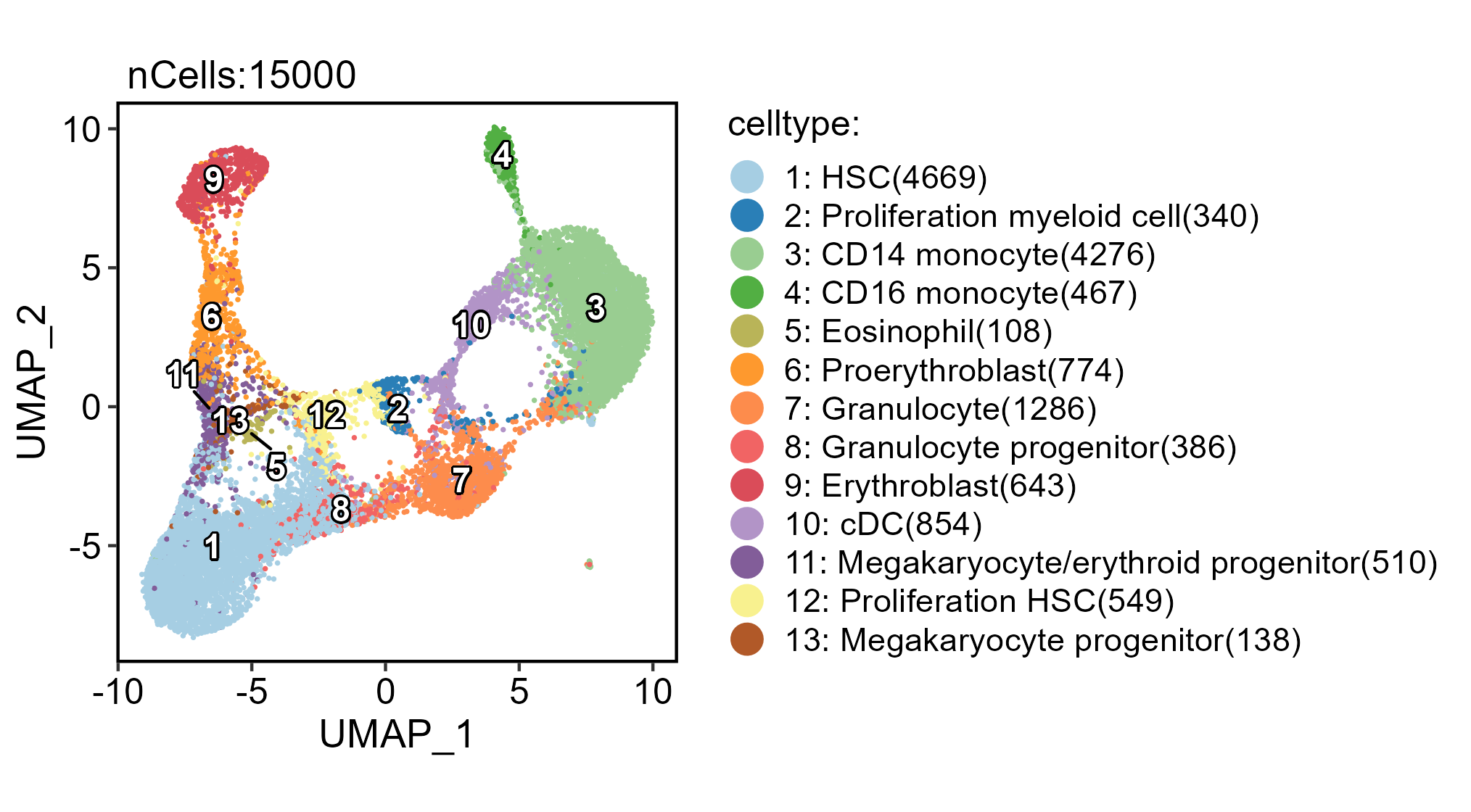
通常情况下,做轨迹分析之前,我们现在已经拥有一个经过质控、降维、并注释好的 seurat 对象,并且你应该对你的细胞轨迹有一个基本的生物学概念。我们简单看看注释的情况:
从下图我们初步可以判断,从生物学角度,HSC 作为发育起点,大致存在 HSC --> Myeloid 和 HSC --> Erythroblast 两个 lineage。
slingshot 接收的是 sce 对象,因此我们首先要构建一个。
在此我们把矩阵提取出来构建,你也可以网上找 seurat 对象直接转换的方法。
# 加载R包
# 首先你需要把这些包安装好
suppressPackageStartupMessages({
library(slingshot)
library(SingleCellExperiment)
library(qs)
library(tidyverse)
library(RColorBrewer)
})
# 加载数据
scobj = qs::qread(file = 'data/01_data_prepare.qs')
我们把 counts 矩阵提取,用于构建 sce 的对象。
# 在这里为了减少计算压力,我们把高变基因提取出来
# 你可以跟我一样从 scale.data 中提取,也可以直接在 seurat 对象中找出来
scale.data <- scobj@assays$RNA@scale.data
scale.gene <- rownames(scale.data)
counts <- scobj@assays$RNA@counts
counts <- counts[scale.gene,]
# 将表达矩阵转换为SingleCellExperiment对象
# 输入需要counts矩阵,否则影响下游分析
sim <- SingleCellExperiment(assays = List(counts = counts))
2. 提取其他有用的信息
为了与前面画图一致,我们把降维坐标和细胞标签也提取并加入 sce 对象。
# umap reduction
umap = scobj@reductions$umap@cell.embeddings
colnames(umap) = c('UMAP-1', 'UMAP-2')
# 将降维的结果添加到SingleCellExperiment对象中
reducedDims(sim) = SimpleList(UMAP = umap)
# metadata
meta = scobj@meta.data
# colData(sim)相当于meta.data,但他不是data.frame格式
# 所以需要分步赋予
colData(sim)$sampleId = meta$sampleId
colData(sim)$celltype = meta$celltype
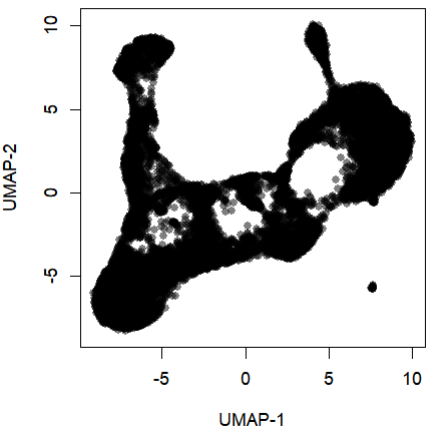
我们可以直接用 plot() 函数画一下看看:
rd = umap
plot(rd, col = rgb(0,0,0,.5), pch=16, asp = 1)
现在,sim 的信息如下:
class: SingleCellExperiment
dim: 1000 15000
metadata(0):
assays(1): counts
rownames(1000): HES4 ISG15 ... MT-ND4L MT-ND6
rowData names(0):
colnames(15000): GGCTGGTAGTGGGCTA-1--GSM3901485
CTACACCTCTCTGTCG-1--GSM3993354 ...
GACGCGTTCCGTAGTA-1--GSM5047372
GGCTCGACATTGGGCC-1--GSM3901492
colData names(2): sampleId celltype
reducedDimNames(1): UMAP
mainExpName: NULL
altExpNames(0):
3. 细胞亚群划分
在这里我们也可以使用自己注释好的细胞作为起点或者终点,但有时候效果并不是那么好,需要多尝试几种方式,以达到满意的效果。slingshot 官方提供了两种方式:高斯混合模型和K-means,如果你有兴趣,可以到官网查看,我在示例数据中也提供了相关的代码。根据个人的经验,使用的分群越精细,效果反而不那么好,轨迹会出现较多重叠的现象,或出现不符合生物学常识的轨迹。
在这里我们使用已经注释好的细胞类型进行轨迹推断。
4. 使用slingshot构建分化谱系并进行拟时推断
在 slingshot 中,我们可以根据我们已有的信息指定拟时序的起点:
如果你没有已知的起点,slingshot 会自己给你拟合一个预测的起点,但通常不太准,或者与实际的时序信息是相反
# 一行命令就可生成,这里计算是比较快的
sim <- slingshot(sim,
clusterLabels = 'celltype', # 选择colData中细胞注释的列名
reducedDim = 'UMAP',
start.clus= "HSC", # 选择起点
end.clus = NULL # 这里我们不指定终点
)
colnames(colData(sim))
# [1] "sampleId" "celltype" "slingshot"
# [4] "slingPseudotime_1" "slingPseudotime_2" "slingPseudotime_3"
# [7] "slingPseudotime_4"
我们可以看到,sim 的 colData 中添加了一些时序信息,每一个 lineage 添加了一列信息,每列信息以时序信息(拟时间)组成,如果细胞对应不在这个 lineage 中,则以 NA 表示。
head(colData(sim)[,4:5])
# DataFrame with 6 rows and 2 columns
# slingPseudotime_1 slingPseudotime_2
# <numeric> <numeric>
# GGCTGGTAGTGGGCTA-1--GSM3901485 1.54205 1.54208
# CTACACCTCTCTGTCG-1--GSM3993354 13.46079 13.44990
# CACATAGTCTGATTCT-1--GSM3901488 NA NA
# GGTGAAGAGTTGGACG-1--ERX4720942 19.42507 NA
# CACCACTAGGTCATCT-1--GSM3396162 NA 22.34963
# GTTACAGCAGATTGCT-1--GSM3901485 2.27809 2.28129
5. 可视化
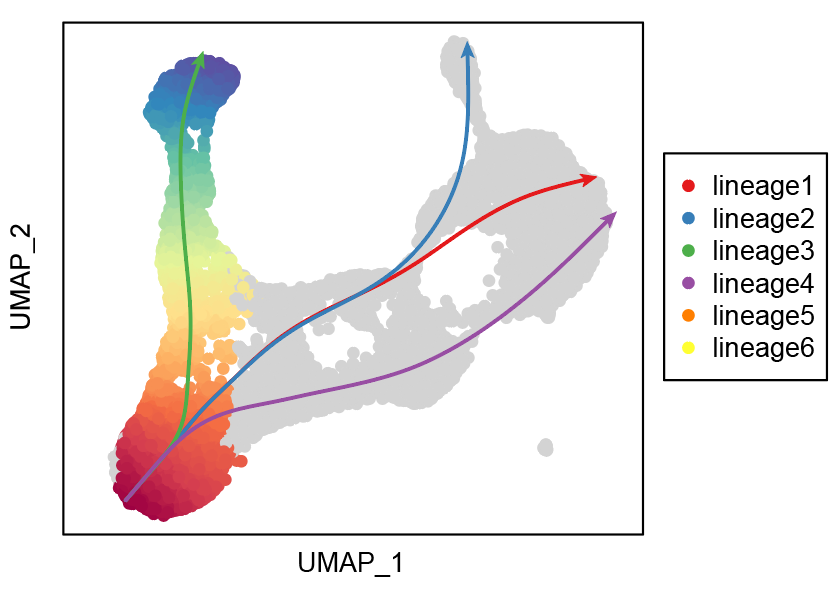
首先我们看看我们各个 lineage 是什么样的,我们使用官方提供的代码
plot(reducedDims(sim)$UMAP, pch=16, asp = 1)
lines(SlingshotDataSet(sim), lwd=2, col=brewer.pal(9,"Set1"))
legend("right",
legend = paste0("lineage",1:6),
col = unique(brewer.pal(6,"Set1")),
inset=0.8,
pch = 16)
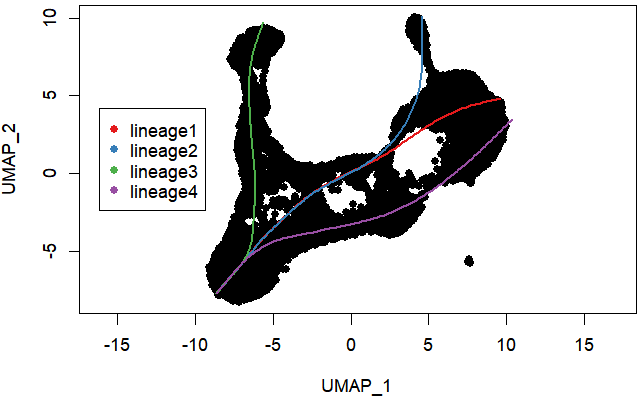
这些 lineage 大致都符合我们先前注释的亚群背后的生物学知识,也有轨迹出现部分重叠,可以设置 slingshot 参数 reweight = FALSE ,让细胞不会在多个轨迹间重复评估。
接下来,假设我们现在对 lineage3 非常感兴趣,我们希望把 lineage2 进行可视化。
首先我们为细胞赋予颜色:
summary(sim$slingPseudotime_2)
# Min. 1st Qu. Median Mean 3rd Qu. Max. NA's
# 0.000 2.207 4.050 6.339 10.041 18.065 9446
# 我们做一个绚丽的渐变色彩虹色
colors <- colorRampPalette(brewer.pal(11,'Spectral')[-6])(100) # 我们把这些颜色变成100个梯度,模拟渐变色
plotcol <- colors[cut(sim$slingPseudotime_3, breaks=100)] # 这里我们用cut函数把 lineage2 分割成100个区间,同一区间的细胞给与同一个颜色
plotcol[is.na(plotcol)] <- "lightgrey" # 不属于 lineage3 的点为NA,我们把他们变成灰色
plotcol
来看看效果:
(但是这些 lineage 数据怎么用 ggplot2 系统绘画,笔者还没找到方法)
# 保存时,把下面的注释运行
# pdf(file = "figures/04_lineage2.pdf",width = 7,height = 5)
plot(reducedDims(sim)$UMAP, col = plotcol, pch=16, asp = 1)
lines(SlingshotDataSet(sim), lwd=2, col=brewer.pal(9,"Set1"))
legend("right",
legend = paste0("lineage",1:6),
col = unique(brewer.pal(6,"Set1")),
inset=0.8,
pch = 16)
# dev.off()
还挺酷的吧~
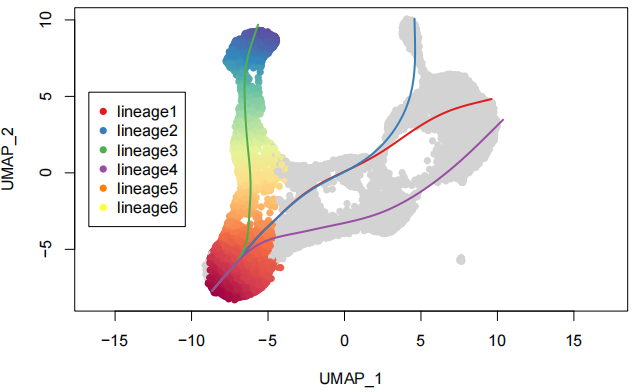
接下来你可以保存成pdf,放在 Adobe Illustrator 中修改一些细节,最后我们的效果如下:
5. tradeSeq 下游分析
这一部分的知识点比较烧脑,我们主要掌握使用的关键点就可以了。
(1)我们对 lineage2 进行下游分析,首先对数据进行预处理。
当我们纳入的细胞类型较少、复杂程度低、轨迹符合生物学背景时,拟合的模型会接近好。当复杂程度较高(如细胞类型混杂),模型会受到大量噪音影响。
# sce对象的colData并不是 data.frame 格式,我们需要重新构建
# 如果你知道更简便的方法,欢迎在评论区提出
coldata <- colData(sim)
coldata <- data.frame(celltype = coldata@listData$celltype,
sampleId = coldata@listData$sampleId,
plotcol = plotcol) # 修改:之前这里是coldata@listData$plotcol,但是读者提醒我前面没有赋值
rownames(coldata) = sim@colData@rownames
# 我们把 lineage3 信息筛选出来
# 细胞 barcodes
filter_cell <- dplyr::filter(coldata, plotcol != "lightgrey")
filter_cell <- rownames(filter_cell)
head(filter_cell)
# 矩阵 counts
counts <- sim@assays@data@listData$counts
filter_counts <- counts[,filter_cell]
filter_counts[1:5,1:5]
(2)tradeSeq 运行较慢,强烈建议随机抽取2000~3000个细胞(不会影响结果的稳定性)
# 随机选取2000个细胞,你可以根据实际情况调整
# 降低计算时间和压力
set.seed(111)
scell <- sample(colnames(filter_counts), size = 2000)
# 最后的到抽样后的矩阵
filter_counts = filter_counts[, scell]
dim(filter_counts)
# 重新将表达矩阵转换为SingleCellExperiment对象
filter_sim <- SingleCellExperiment(assays = List(counts = filter_counts))
# colData
filter_coldata = colData(sim)[scell, 1:3]
filter_sim@colData = filter_coldata
# reduction
rd = reducedDim(sim)
filter_rd <- rd[scell,]
reducedDims(filter_sim) <- SimpleList(UMAP = filter_rd)
(3)由于重新构建sim,之前的轨迹信息我们需要重新做一遍
这里我们需要换一个细胞分类信息,还记得在前面《细胞分类》这部分讲过,当纳入slingshot的分类信息越多越复杂,就更有可能导致不理想的结果出现,如多个轨迹重叠。
我们这里已经把lineage3选出来,但其成分仍然非常复杂,如果单纯以目前注释的celltype进行分析,则会出现很多杂乱无章的信息。我们需要换一种方式更完美的还原lineage3,在这类我推荐前面提到过的K-Means方式。
# K-Means
set.seed(111)
cl <- kmeans(filter_rd, centers = 4)$cluster # 可以指定分群数量
head(cl)
colData(filter_sim)$kmeans <- cl
## 可视化聚类分群的结果
library(RColorBrewer)
mycolors = brewer.pal(4,"Set1") # colors
plt_k = data.frame(filter_rd,
kmeans_clusters = factor(cl, levels = sort(unique(cl))))
ggplot(plt_k, aes(UMAP_1, UMAP_2))+
geom_point(aes(color = kmeans_clusters), size = .5)+
scale_color_manual(values = mycolors)+
theme_bw()+
theme(panel.grid = element_blank(),
axis.text = element_blank(),
axis.ticks = element_blank())+
xlab('UMAP_1')+
ylab('UMAP_2')+
guides(color = guide_legend(override.aes = list(size = 5)) # 调整图例展示的点大小
)
K-Means 可以理解为按“区域划分”,可以指定 K-Means 分割的区域数量,从而我们可以有效地控制整个轨迹地走向。否则细胞亚群可能东一块、西一块,对轨迹分析影响很大。

# 使用 K-Means 划分的亚群进行轨迹推断
filter_sim <- slingshot(filter_sim,
clusterLabels = 'kmeans',
reducedDim = 'UMAP',
start.clus= "4", # 指定起点
end.clus = '3' # 指定终点
)
head(colnames(colData(filter_sim)))
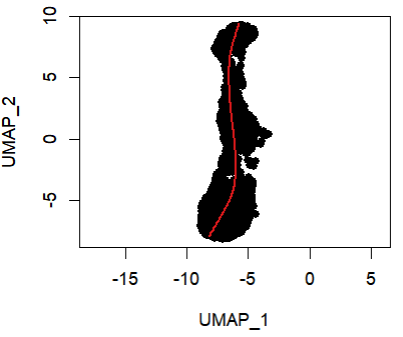
plot(reducedDims(filter_sim)$UMAP, pch=16, asp = 1)
lines(SlingshotDataSet(filter_sim), lwd=2, col=mycolors)
基本复现了 lineage3 的轨迹,如果你喜欢探索,可以尝试直接用 HSC 作为起点,Erythroblast 为终点。就会明白为什么小编会推荐你用 K-Means(T_T)。
(4)使用tradeSeq软件包中的负二项式广义加性模型(NB-GAM)进行拟合
# BiocManager::install("tradeSeq")
# 如果你的安装有问题,可以尝试手动安装,我也在示例数据压缩包中放了最新的tradeSeq安装包
library(tradeSeq)
# Fit negative binomial model
counts <- filter_sim@assays@data$counts
crv <- SlingshotDataSet(filter_sim)
# 在使用NB-GAM模型之前,需要确定用于构建模型的基函数的数量。
# 这些基函数被称为节点(knots),它们在模型的拟合过程中起到关键作用。
# 使用evaluateK函数。该函数会运行一段时间
# 2k cells ~16min(如果你的轨迹较多的话,时间更长)
# 运行之后会自动出现图
set.seed(111)
icMat <- evaluateK(counts = counts,
sds = crv,
k = 3:10, # no more than 12
nGenes = 200, # 每个细胞纳入分析的基因数量,默认是500,这里为了节省示例计算时间
verbose = T)
# |++++++++++++++++++++++++++++++++++++++++++++++++++| 100% elapsed=02m 11s
# |++++++++++++++++++++++++++++++++++++++++++++++++++| 100% elapsed=01m 45s
# |++++++++++++++++++++++++++++++++++++++++++++++++++| 100% elapsed=02m 24s
# |++++++++++++++++++++++++++++++++++++++++++++++++++| 100% elapsed=02m 01s
# |++++++++++++++++++++++++++++++++++++++++++++++++++| 100% elapsed=02m 06s
# |++++++++++++++++++++++++++++++++++++++++++++++++++| 100% elapsed=01m 34s
# |++++++++++++++++++++++++++++++++++++++++++++++++++| 100% elapsed=02m 52s
# |++++++++++++++++++++++++++++++++++++++++++++++++++| 100% elapsed=01m 21s
我们需要根据下图来确定 nknots,详细解释见下面链接:
https://statomics.github.io/tradeSeq/articles/fitGAM.html
如果你只想快速确定,那么我们只需要看中间的两个图,我们找到快速下降曲线的转折点;如果这个点小于6,则选择对应的数字;如果这个点大于等于6,则选择6。
下图中,我们可以看到曲线变平滑的转折点是6之后,则我们选择 nknots = 6。
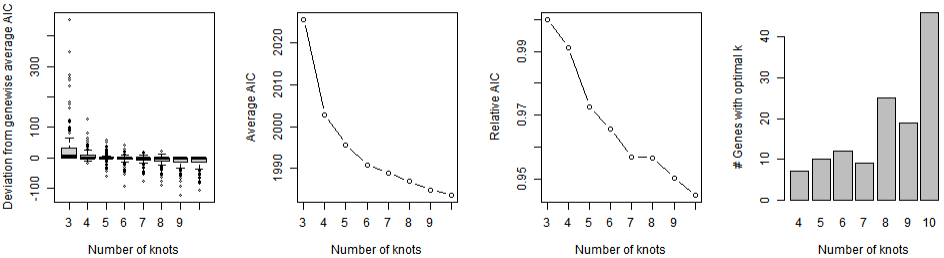
# we pick nknots = 6.
set.seed(111)
pseudotime <- slingPseudotime(crv, na = FALSE)
cellWeights <- slingCurveWeights(crv)
# fit negative binomial GAM
# 2k cells ~13 min
# system.time()这个函数可以计算运行时间
system.time({
sce <- fitGAM(counts = counts,
pseudotime = pseudotime,
cellWeights = cellWeights,
nknots = 6,
verbose = FALSE)
})
table(rowData(sce)$tradeSeq$converged)
# FALSE TRUE
# 171 829
该模型可能难以适用于某些基因,因此拟合过程可能不会收敛到一个数据集中的所有基因,特别是在具有复杂模式和/或许多谱系的数据集中。您可以检查每个基因的收敛性,如下所示,其中
TRUE值对应于收敛模型拟合,而FALSE值对应于未能完全收敛的模型。
FALSE值指的是模型未能完全收敛,即这些基因的拟合过程可能存在困难,并不意味着这些基因与轨迹变化无关。模型未能完全收敛可能是因为基因的特征非常复杂或数据集中包含许多细胞亚群,导致模型难以准确地拟合。
(5)探索基因表达与拟时序的相关性
# test for dynamic expression
assoRes <- associationTest(sce)
head(assoRes)
# waldStat df pvalue meanLogFC
# HES4 4.495296 5 4.805131e-01 4.1062521
# ISG15 46.567234 5 6.960814e-09 0.8088061
# MXRA8 NA NA NA 0.1996149
# SMIM1 1292.662958 5 0.000000e+00 1.7751929
# RBP7 1.741292 5 8.836572e-01 244.3457299
# PLA2G2A NA NA NA 0.1996149
associationTest函数应用于sce对象,用于对每个基因进行检验以确定其在拟时中是否存在显著的关联性。结果保存在assoRes变量中,并通过打印前几行来查看。输出结果包含了以下列:
waldStat:Wald统计量,用于衡量基因表达与拟时的关联程度。df:自由度,用于衡量统计推断的准确性。pvalue:显著性水平(p值),用于评估基因表达与拟时之间的关联是否具有统计学意义。meanLogFC:均值对数倍数差异(Fold Change),表示基因在不同拟时阶段的平均表达差异。
请注意,输出结果中一些基因的值为NA是因为它们未能通过检验或计算相关的统计指标。只有具有非NA值的基因才能提供关于基因表达与拟时之间关联性的信息。
可以关注以下两个方面:
pvalue列:检查每个基因的p值,如果p值小于设定的显著性水平(通常为0.05),则可以得出结论认为基因表达与拟时之间存在显著的关联性。meanLogFC列:观察基因的均值对数倍数差异,正值表示基因在拟时上升的过程中表达增加,负值表示基因在拟时上升的过程中表达下降。
(6)寻找与起止点相关性最高的基因
# Discovering progenitor marker genes
startRes <- startVsEndTest(sce)
head(startRes)
# waldStat df pvalue logFClineage1
# HES4 1.011937e+00 1 3.144393e-01 -10.1403413
# ISG15 2.027631e+01 1 6.702522e-06 1.6526103
# MXRA8 7.526157e-15 1 9.999999e-01 -0.3659606
# SMIM1 1.267845e+02 1 0.000000e+00 3.3647451
# RBP7 3.080566e-01 1 5.788752e-01 332.3079668
# PLA2G2A 7.526157e-15 1 9.999999e-01 -0.3659606
startVsEndTest使用Wald检验来评估零假设,即平滑器起始点(祖细胞群)的平均表达是否等于平滑器终点(分化细胞群)的平均表达。该检验基本上涉及每个亚群的两个平滑器系数的比较。默认情况下,startVsEndTest函数在所有亚群中执行全局检验(即同时比较起始点和终点),但你也可以通过设置lineages=TRUE来单独评估每个lineage。
在这个例子中,我们已经把lineage3单独挑选出来运算,并根据K-Means复现了轨迹,因此实际上我们只有一个lineage。所以(5)和(6)的运行结果是相似的。如果你有多个lineage,应该在这一步加上lineages=TRUE。但不建议同时有多个轨迹。
# 按相关性进行排序
oStart <- order(startRes$waldStat, decreasing = TRUE)
# 挑选相关性最强的基因,并可视化
sigGeneStart <- names(sce)[oStart[1]]
plotSmoothers(sce, counts, gene = sigGeneStart)

我们也可以用UMAP图展示
plotGeneCount(crv, counts, gene = sigGeneStart)

(7)美化基因表达量变化图
上述两个基因表达量变化图是通过 ggplot2 系统绘制的,我们可以通过添加一些自定义参数来美化他。
我们提取需要的数据,并构建成清洁数据框。
# 取celltpye和配色信息
coldata <- data.frame(celltype = sim@colData$celltype,
plotcol = sim@colData$plotcol)
rownames(coldata) = colnames(sim)
# 把sce中的3000个细胞对应信息取出
filter_coldata <- coldata[colnames(sce),]
# 添加拟时序信息
filter_coldata$Pseudotime = sce$crv$pseudotime.Lineage1
# top6 genes
top6 <- names(sce)[oStart[1:6]]
top6_exp = sce@assays@data$counts[top6,]
top6_exp = log2(top6_exp + 1) %>% t()
# 获得最终的清洁数据
plt_data = cbind(filter_coldata, top6_exp)
colnames(plt_data)
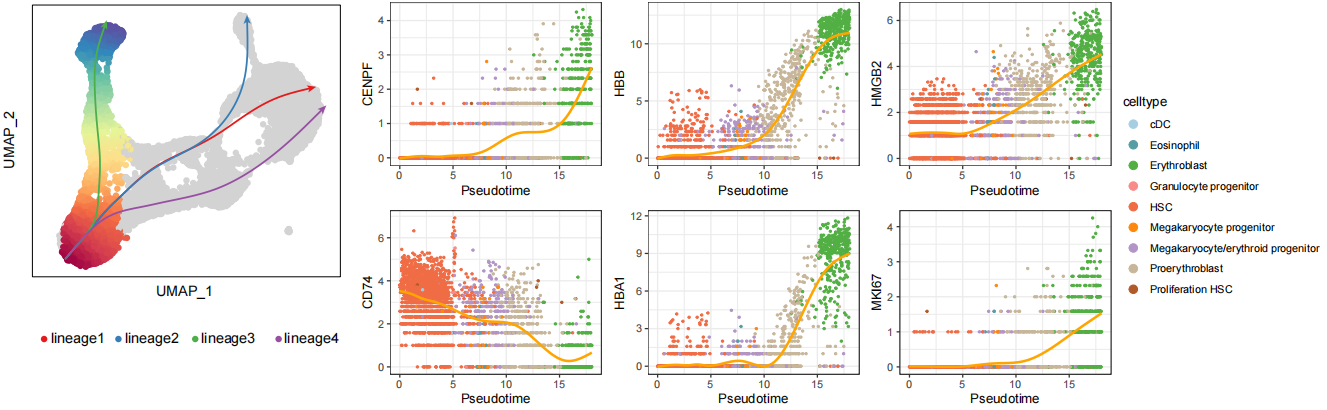
# [1] "celltype" "plotcol" "Pseudotime" "CENPF" "HBB"
# [6] "HMGB2" "CD74" "HBA1" "MKI67"
画图
library(ggplot2)
library(ggsci)
library(ggpubr)
library(RColorBrewer)
getPalette = colorRampPalette(brewer.pal(12, "Paired"))
mycolors = getPalette(length(unique(plt_data$celltype)))
# 为了拼图美观,我们把legend隐藏掉
plt_list = list()
for (gene in top6) {
print(gene)
p = ggscatter(data = plt_data,
x = 'Pseudotime',
y = gene,
color = 'celltype',
size = 0.6)+
geom_smooth(se = F, color = 'orange')+
theme_bw()+
scale_color_manual(values = mycolors)+
theme(legend.position = 'none')
plt_list[[gene]] = p
}
library(patchwork)
wrap_plots(plt_list)
ggsave(filename = 'figures/05_top6_genes.pdf', width = 9, height = 5)

# 这里单独save一张有legend的图
gene = 'HBB'
p_test = ggscatter(data = plt_data,
x = 'Pseudotime',
y = gene,
color = 'celltype',
size = 0.6)+
geom_smooth(se = F, color = 'orange')+
theme_bw()+
scale_color_manual(values = mycolors)+
theme(legend.position = 'right')+
guides(color = guide_legend(ncol = 1, # 将图例分为两列显示
override.aes = list(size = 3)) ) # 调整图例展示的点大小
p_test
ggsave(plot = p_test, filename = 'figures/05_HBB_legend.pdf', width = 5, height = 5)
(8)美化拼图
最后,我们把上面的元素用 AI 软件做个拼图

总结
那么今天分享就到这里了。整体而言,相比于 Monocle 工具,笔者认为 slingshot 是一个更加轻量的工具,他的计算速度较快,步骤较少,数据提取容易,既可以用官方的可视化方式,也可以基于 ggplot 系统进行优化,是一个不错的工具。除了上述展示方式以外,你还可以将这些基因进行富集分析,并用UCell 等打分工具对细胞进行平分,可视化到 UMAP 图上。官方的教程中还有许多其他展示方式,有兴趣自己翻阅吧!

 浙公网安备 33010602011771号
浙公网安备 33010602011771号