文献阅读 | Parent-independent genotyping for constructing an ultrahigh-density linkage map based on population sequencing
Xie, W., Feng, Q., Yu, H., Huang, X., Zhao, Q., Xing, Y., Yu, S., Han, B., & Zhang, Q. (2010). Parent-independent genotyping for constructing an ultrahigh-density linkage map based on population sequencing. Proceedings of the National Academy of Sciences of the United States of America, 107(23), 10578–10583.
在这篇2010年的文章中,作者报道了一种在重组自交系上利用低覆盖率测序构建由SNP组成的超高密度(ultrahigh-density)连锁图的方法。在方法中,作者首先采用最大简约推断法,识别了所有可能有助于构建亲本基因型草图的SNP,以最大化利用群体中发现的SNP信息。随后,通过划定窗口重采样和贝叶斯推断,作者进一步筛选出了高质量SNP。再利用HMM模型,借助高质量SNP对作图群体(mapping population)中的株系及逆行那个基因分型。
在238个重组自交系、平均每株系0.05x测序下,作者构建了一个由优质SNP小区间(bins of high-quality SNPs)组成的超高密度连锁图谱,并利用该图谱将一个粒宽数量性状(GW5)成功定位到200kb的区间中。该方法普遍适用于低覆盖序列数据的遗传图谱构建。
对RILs进行测序,并鉴定在无亲本序列下鉴定潜在双亲SNP。
A total of 238 RILs developed from the cross between Zhenshan 97 and Minghui 63 were resequenced with an Illumina Genome Analyzer II using the bar-coded multiplexed sequencing approach.
(细节略)经测序,每自交系获得了约20.6 Mb sequence,相当于∼0.055-fold genome coverage。
作者将所有定位质量不低于40的RILs的定位序列(mapped sequences)进行了合并(merge),以得到每个位点的核苷酸信息。由于一个真实SNP位点的核苷酸应该是双等位的,所以根据它们的双峰分布(bimodal distribution)来确定潜在的SNP位点和两个等位核苷酸。作者在chromosome 5上共鉴定出了15795个双亲SNP位点,相当于每1.9kb一个SNP位点。筛除潜在的假阳性SNP后,共获得了209240个SNP位点,使得RIL中的SNP平均密度为3/100kb。
为了评估上述方法的准确率,作者从所有RILs中鉴定了不同区间的共识序列。同时,将双亲在同样实验条件下测定了约0.032x。通过比较,发现Zhenshan 97中有3.18%个位点与推测不一致。
利用重组的最大简约性推断亲本基因型
要构建遗传连锁图谱,必须了解亲本的基因型。在高通量测序时代,如果序列深度较低,亲本序列信息提供的有用信息很少,不能作为构建图谱时亲本SNP基因型的参考。作者提出了一种利用最大简约重组(maximum parsimony of recombination,MPR)原理推断亲本基因型的方法。
对于一组给定的RIL基因型,如果对重组事件的数量没有限制的话,“推断的亲本基因型”将有许多可能性。通过MPR原理,假设亲本基因型是产生重组事件数最少的RIL基因型。
简单起见,该想法用一个由5个SNP位点和6个RIL组成的假设数据集来说明(矩阵a,图1)。低覆盖率数据集的一个共同特征是,它在很大一部分SNP位点(显示为空白单元格)包含缺失的数据。将矩阵A中5个snp(行)和6个RIL(列)的双等位基因状态提取到矩阵B中5行2列(A和B)。假设a列中的等位基因均来自亲本1,b列中的等位基因均来自亲本2,则需要6个重组事件来产生6个RIL的基因型(矩阵C)。
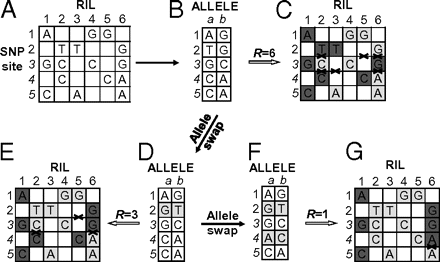
Fig. 1.
An example of inferring parental genotypes based on the principle of maximum parsimony of recombination (MPR). Different background colors represent different genotypes in matrices C, E, and G, whereas crosses indicate the recombination breakpoints between different genotypes. See text for details.
为找到能通过最少的重组次数产生6个RIL的两个亲本基因型,作者在a列和B列之间每次交换一个等位基因,对假定的亲本基因型(矩阵B)进行迭代地逐步扰动,并且所得的“假定基因型矩阵”(矩阵D)再次用于推断重组事件:在这个过程中,交换第二个SNP位点的等位基因能将重组事件从6个减少到3个,如矩阵E所示。因此,在矩阵中固定这些等位基因,并继续等位基因交换过程。最终,在第四个SNP位点(matrix F)进行的另一次交换可以产生只有一个重组事件的RIL基因型,这是这个数据集可能出现的最小数目。因此,接受矩阵F作为亲本基因型。
(该方法是MPR假设下的贪心算法)
这个过程可以扩展到一个更一般的情况,即由一个局部基因组区域(窗口)中n个假定的SNP位点和m个RILs(或其他类型的作图群体)组成的数据集。唯一的例外是,交换等位基因的SNP位点的数量(称为交换的步长)不一定是一次一个,尽管它总是从一个开始。如果一次交换一个等位基因不减少重组事件(R)的数量,则步长可以一次增加一个SNP位点,直到R可以减少或步长达到定义的最大步长sm。这种迭代交换将继续,直到R不能进一步减少。
利用MPR方法,作者根据RILs的低覆盖序列推断亲本基因型。在该过程中,首先将染色体分成数百个小窗口,每窗口包括50个SNP(图2A)。
根据Zhenshan 97序列(称为ZS SNPs)恢复的SNP位点进行窗口合并,以确保每个窗口中至少包含10个ZS SNPs(图2B)。每个窗口内使用MPR过程利用所有RIL的SNP数据来预测亲本的基因型。由于已知10个ZS单核苷酸多态性的Zhenshan 97基因型,尽管有一定的错误率,但如果有必要,该窗口中其他单核苷酸多态性的基因型将被指定为Zhenshan 97≥80%的ZS-SNPs基因型与预测一致。最后,根据每个窗口中的ZS单核苷酸多态性,对来自不同窗口的基因型调用进行组合,为两个亲本(默认情况下,如果一个亲本被确认,另一种可能将是另一个亲本)、Zhenshan 97和Minghui 63(图2C)生成“基因型草图”。
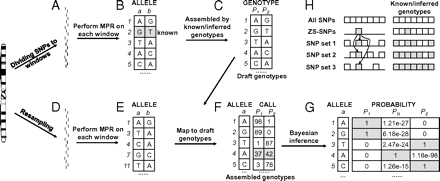
Fig. 2.
The scheme for global inference of parental genotypes. (A–C) Procedures for inferring the draft genotypes of the two parents P1 and P2 assisted by low-coverage parental sequences of one parent (see text for details). (D–G) Refining the SNPs of the inferred parental genotypes, including: (D and E) inferring the parental genotypes using permutation by resampling of SNPs according to individual RILs (see text for details), and (F and G) validating the SNPs by posterior probabilities (Bayesian inference). The number of genotype calls for an SNP site resulting from the permutations was recorded (F), and the probability for the assignment of the draft genotype was calculated (G), whereby the genotype is assigned by the highest posterior probability of P1, PN, and P2 at the given SNP site. It is also seen that the posterior probability of PN of SNPs at the fourth site (in gray) is the highest, and thus this site is regarded as a low-quality SNP site. (H) Assembling genotype calls from different windows aided by low-coverage parental sequences.
通过重采样和贝叶斯推理来细化亲本基因型,以去除低质量的SNPs。
在上述推测的基因型草案中可能会出现不同来源的错误。首先,Zhenshan 97的测序错误会降低ZS-SNPs的可靠性。第二,RILs中的一些SNP位点虽然是双等位基因,但由于测序错误或同源区域的错误比对,其质量可能不高。第三,偶然事件也可能导致MPR方法推断的亲本基因型的错误。
为了验证推断的基因型,作者设计了一种排列置换法,包括局部重采样和贝叶斯推断,以筛选出低质量的SNPs并细化亲本基因型预测(图2d-G)。
在这个排列过程中,作者沿着染色体排列了所有的单核苷酸多态性。排列基于238个RIL的划分,因此由238个步骤组成。在第一步中,在第一个RIL(R001)中检测到的前100个SNPs中随机抽取50个,不进行替换,并使用MPR方法进行分析以获得基于50个SNP的亲本单倍型推断。如果≥80%的单核苷酸多态性符合亲本基因型草图,则分配对应样本的单倍型。然后,包括在第一步中的50个未被抽样的前100个snp中再次随机取样50个而不替换,并且使用MPR方法进行分析以获得如上所述的两个亲本基因型的另一推断。重复这个过程,直到R001中出现的所有单核苷酸多态性都被用于推断,从而在每个SNP位点推断出每个亲本的SNP基因型。
整个排列过程重复了20次。因此,如果对238个RIL中的10个进行核苷酸测序,将获得该SNP的200个基因型calls。
接着,作者在两个条件下评价MPR方法的排列数和错误率对可靠SNP保留率的影响:通过后验概率(贝叶斯推断)丢弃低质量的SNP位点,以及去除次要基因型数不小于排列数的位点。
结果表明,根据排列的数量和错误率(从0.05到10%),保留了76.7–84.3%的假定SNPs(图S2),并且假设MPR的错误率较小,则剩余SNPs较少。经过10次排列后,SNPs的数量保持稳定。采用MPR方法的保守错误率为0.05%,通过10次排列共鉴定出13227个单核苷酸多态性。
作者还将这些单核苷酸多态性与排列产生的13227个高质量单核苷酸多态性进行了比较。结果发现,94.1%(970/1031)的劣质SNP被置换去除,因此不包括在精简集中,而98.7%(11792/11948)的高质量SNP位点保留在精简集中。所有11792个位点的亲本基因型调用与测序结果一致,证实了本研究所用方法在SNP鉴定和亲本基因型推断方面的可靠性。值得注意的是,在从RIL序列中鉴定的13227个SNP中,有10.8%(1435)没有被鉴定为来自深度测序的SNP,主要是因为没有足够的来自一个或两个亲本的SNP位点的序列来满足SNP鉴定的要求。
利用隐马尔可夫模型对RIL进行基因分型。
直接使用低覆盖率序列进行RIL基因分型有两大困难。首先,RILs原始序列的错误率较高(估计为3.18%),这将影响单个SNPs基因分型的准确性。其次,一个杂合子的SNP位点的两个等位基因中只有一个可以被观察到,而位于杂合子区域的相邻SNP将以两种基因型的混合出现,从而导致基因型calling困难。为了克服这些困难,作者将染色体上的单核苷酸多态性基因型作为马尔可夫链来处理。一个简单的隐马尔可夫模型(HMM)用于根据局部区域的SNP可用信息估计RIL最可能的潜在SNP基因型(图3)。
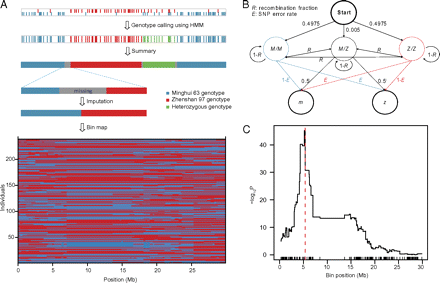
Fig. 3.
RIL genotyping, bin map construction, and mapping of QTLs controlling grain width. (A) Procedures of RIL genotyping and bin map construction. The lower panel shows the bin map of 238 rice RILs on chromosome 5. (B) Probabilistic parameters of the hidden Markov model used to genotype RILs. (C) Mapping curve of QTLs controlling grain width on chromosome 5. A simple Student's t test was used to locate bins associated with grain width. The x axis is the position of bins along the chromosome and the rugs on the x axis represent the borders of bins. The y axis shows the log10-transformed P values resulting from the t test, which represents the degree of association between grain width and bins.
HMM由两个观察值(每个SNP的等位基因,表示为m和z)(图3B)和三个隐藏状态构成,对应于两个纯合子(表示为M/M 和Z/Z)和杂合子(M/Z)。作者从RIL群体中M/M:M/Z:Z/Z三种基因型的预期比例49.75:0.50:49.75开始。由于多重测序的错误率估计为3.18%,只有与SNP位点上的替代核苷酸匹配的序列错误才可能导致不正确的基因型分配(1/3的机会),因此计算出特定SNP位点上RIL基因型调用的错误率为1.06%。在通过计算定义了转移概率和发射概率(详见材料和方法)之后,Viterbi算法被用来确定RILs最可能的潜在基因型。
HMM处理结果中,三种基因型M/M:M/Z:Z/Z的比例为49.16:0.32:50.52。作者使用这个比率再次执行HMM来进行基因型calling,结果是一致的,因此收敛到这个比率。
在随后的分析中,作者将所有杂合位点(0.34%)mask为缺失数据。将具有相同基因型的连续SNP位点集中到块中,并假设在两个不同基因型块之间的过渡处有一个断点。长度小于250kb且序列snp数小于5的片段被掩盖为缺失数据,以避免假双重组。在238个RIL中,5号染色体共鉴定出600个断点。
Constructing the Bin Map and Evaluation by QTL Analysis.
在进一步的处理中,将两个不同基因型区之间的转换区域的基因型设置为缺失数据,并使用R/qtl包进行填充。在两个相邻区块边界共分离的标记被集中为一个bin。在整个RIL群体中合并相同基因型的相邻bin后,获得了238个RIL在5号染色体上总共143个重组库的骨架库图(图3A)。重组子的平均物理长度为208.5kb,从14.0kb到5.17mb不等。

 浙公网安备 33010602011771号
浙公网安备 33010602011771号