文献阅读 | Network medicine framework for identifying drug-repurposing opportunities for COVID-19
Gysi, Deisy Morselli, et al. "Network medicine framework for identifying drug-repurposing opportunities for COVID-19." Proceedings of the National Academy of Sciences 118.19 (2021). https://doi.org/10.1073/pnas.2025581118
作者部署了依赖于人工智能、网络扩散(network diffusion)和网络接近性(network proximity)的算法,对6340种药物进行抗SARS-CoV-2预期疗效的排名。作者挑选了918种基本药物进行了实验验证,以评估现有的药物重利用方法学的表现,并使用一个共识算法(consensus algorithm)来增加预测准确率。最后,作者选出了6种降低病毒感染的药物,其中四种可以治疗COVID-19,提出了治疗新冠的新方法。
作者认为,这一策略具有超出COVID-19的意义,使我们能够识别被忽视疾病替代药物的候选药物。
在试验验证中,作者发现,没有一个单一的预测算法可以在所有数据集和指标之间提供一致可靠的结果。这一结果促使作者开发了一种多模式技术,该技术融合了所有算法的预测,并发现不同的预测方法之间的共识始终超过最佳单个管道的性能。作者在人体细胞中筛选了排名最高的药物,获得了62%的成功率,而非引导性筛选的命中率为0.8%。作者还发现,成功减少病毒感染的77种药物中有76种并不结合SARS-CoV-2靶向的蛋白质,这表明这些依赖于网络机制的网络药物无法通过 docking-based strategies 被发现。
COVID-19指疾病;SARS-CoV-2指病毒。
Pipline
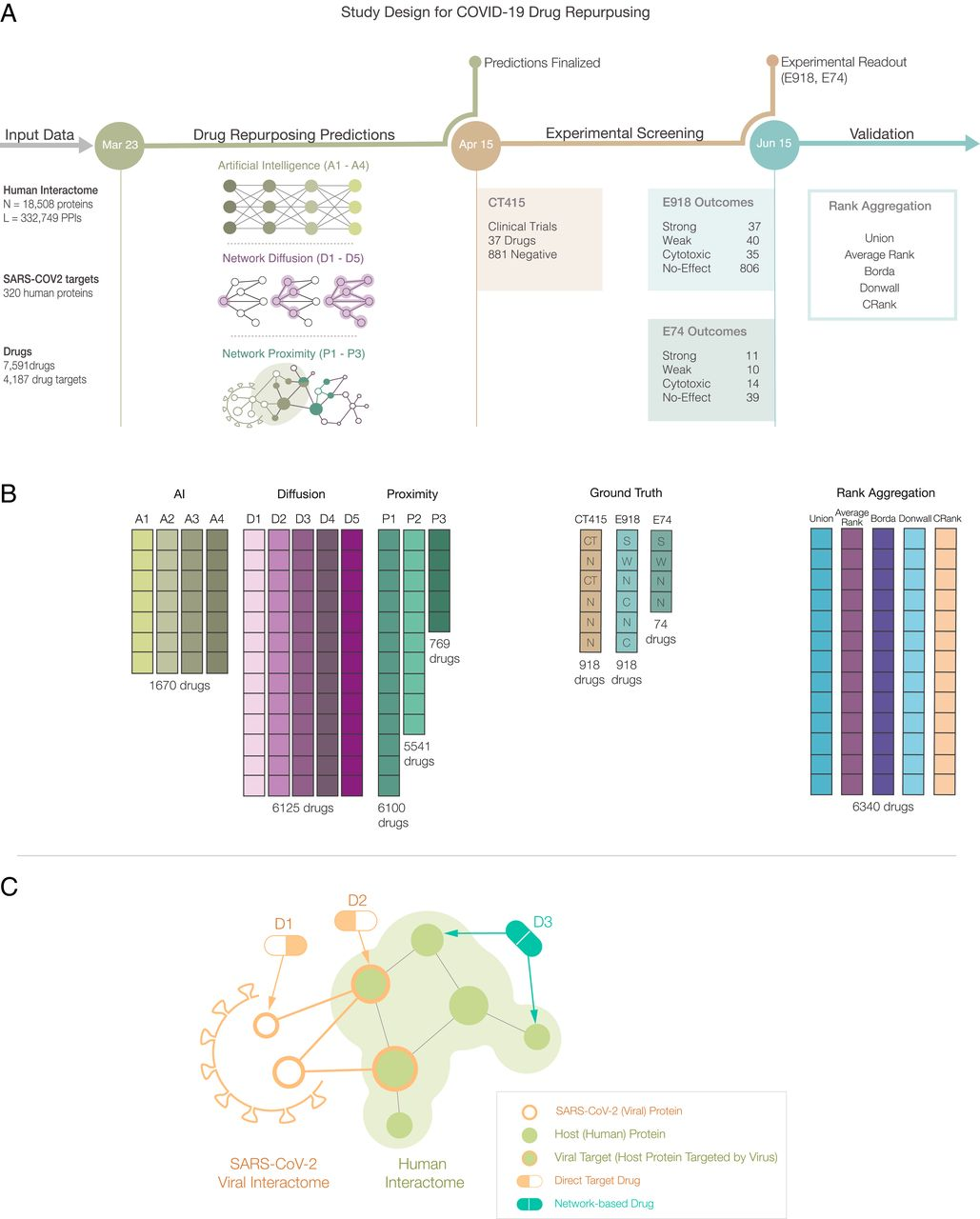
Fig. 1.
Network medicine framework for drug repurposing. (A) Study design and timeline. Following the publication of host–pathogen PPIs (March 23, 2020), we implemented three drug-repurposing algorithms, relying on AI (A1 to A4), network diffusion (D1 to D5), and proximity (P1 to P3), together resulting in 12 predictive ranking lists (pipelines, shown in B). Each pipeline offers predictions for a different number of drugs that were frozen on April 15, 2020. We then identified 918 drugs for which all pipelines but P3 offered predictions, and experimentally validated their effect on the virus in VeroE6 cells. The experimental (E918, E74) and clinical trial lists C415 offered the ground truth for validation and rank aggregation. (C) Direct target drugs bind either to a viral protein (D1) or to a host protein target of the viral proteins (D2). Network drugs (D3), in contrast, bind to the host proteins and limit viral activity by perturbing the host subcellular network.
Results
基于网络的药物再利用
为了搜索与COVID-19分子机制有重合的疾病,作者首先将已有实验验证过的332个SARS-CoV-2宿主靶蛋白定位到人类相互作用组(human interactome,18508个人类蛋白的332749个成对结合相互作用关系)中,并在一个大的连接组件(a large connected component)中发现332个病毒靶蛋白中的208个(图2B),暗示SARS-CoV-2靶蛋白聚集在同一网络附近。
接着,作者基于网络使用\(S_{vd}\)度量了蛋白与229种疾病关联性,并寻找\(S_{vd}>0\)的所有疾病,意味着COVID-19疾病模块不会与与任何单一疾病相关的疾病蛋白直接重叠。换句话说,潜在的COVID-19治疗方案不能从已经批准用于某种特定疾病的疗法中获取。它主张使用一种基于网络的策略,该策略可以识别可重复使用的药物而无需考虑已确定的疾病指征。
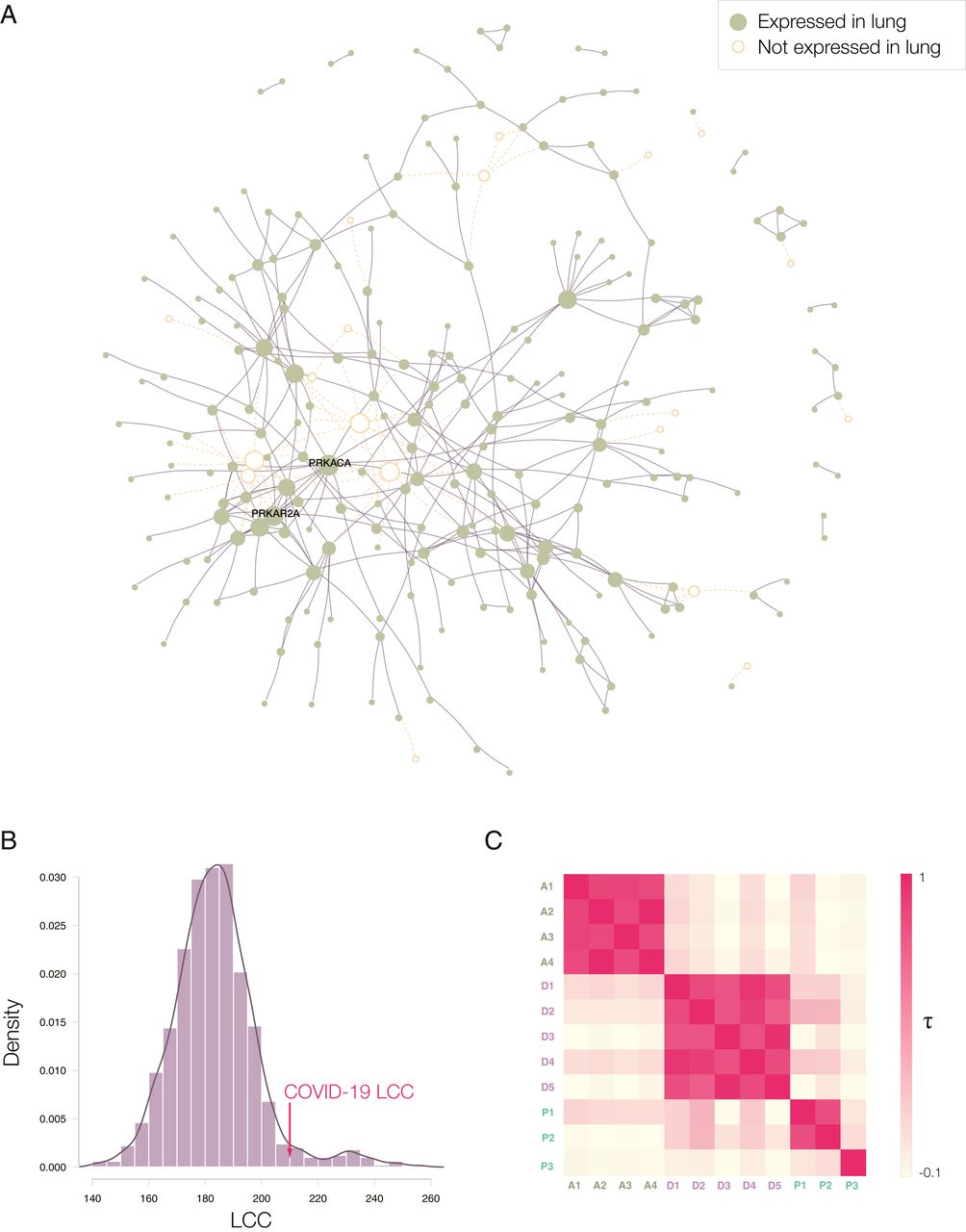
Fig. 2.
COVID-19 disease module. (A) Proteins targeted by SARS-CoV-2 are not distributed randomly in the human interactome, but form a large connected component (LCC) consisting of 208 proteins, and multiple small subgraphs, shown in the figure. Almost all proteins in SARS-CoV-2 LCC are also expressed in the lung tissue, potentially explaining the effectiveness of the virus in causing pulmonary manifestations of the disease. (B) The random expectation of the LCC size indicates that the observed COVID-19 LCC, whose size is indicated by the red arrow, is larger than expected by chance (z-score = 1.65). (C) Heatmap of the Kendall τ statistic showing that the ranking list predicted by the different methods (A, D, and P) are not correlated. We observe, however, high correlations among the individual ranking list predicted by the same predictive method.
作者使用了三种竞争性的网络重用方法(图1B),其中基于AI的有4种pipline(A1-A4),基于扩散算法的有5种(D1-D5),基于临近算法的有3种(P1-P3)。三种算法间的弱相关性表明这些方法从网络中提取到了补充信息(complementary information)。
药物再利用pipline的实验和临床验证。
作者使用基于3种算法的共12个pipline预测了Drugbank种6340种药物对SARS-CoV-2的预期疗效。由于不同的管道成功预测了不同的药物子集,因此作者确定了918种药物。对于这些药物,作者在VeroE6细胞上进行了对SARS-CoV-2功效的比较。在918种药物中,有806种对病毒感染性没有可检测到的影响(N,占被检测清单的87.8%);35种对宿主细胞有细胞毒性(C);37种具有强效作用(S),在很宽的浓度范围内都具有活性;40种对病毒的作用较弱(W)(图3A)。由于预测流程无法提供有关体内效应大小的指示,因此作者将对病毒有强或弱作用的药物定义为阳性药物(S&W,77种药物)(表1),而未能检测到作用的定义为阴性药物(N,806种药物)。
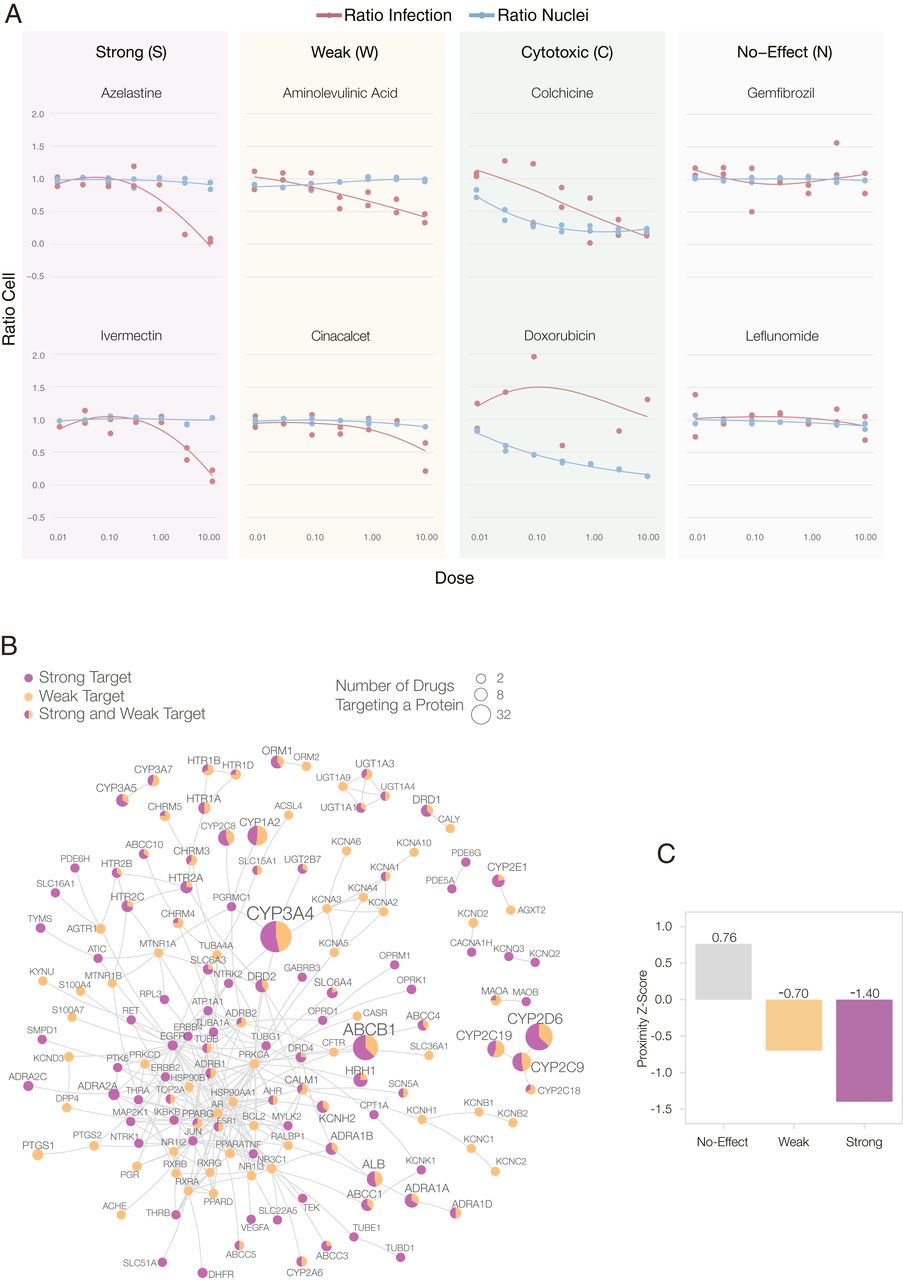
Fig. 3.
Experimental outcomes and network origins. (A) Examples of dose–response curves for eight of the 918 experimentally validated drugs, illustrating the four observed outcomes (S, W, C, and N). VeroE6 cells were challenged in vitro with SARS-CoV-2 virus and treated with the drug over a range of doses (from 8 nM to 8 µM). A two-step drug-response model was used to classify each drug into S, W, C, or N categories, according to their response to the drug in different doses and cell and viral reduction. (B) The subnetwork formed by the targets of the 77 S&W drugs within the interactome. The link corresponds to binding interactions. Purple proteins are targeted by S drugs only; orange by W drugs only; proteins targeted by both S&W drugs are shown as pie charts, proportional to the number of targets in each category. (C) The targets of N drugs have a positive proximity z-score to the COVID-19 module, meaning they are further from the COVID-19 module than random expectation. In contrast, the targets of S&W drugs are more proximal (closer) to the COVID-19 module than expected by change, suggesting that their COVID-19 vicinity contribute to their ability to alter the virus’s ability to infect the cells.
2020年4月15日,作者又扫描了clinicaltrials.gov,识别了134个COVID-19临床试验(CT415 dataset)中的67种药物。为了比较各个数据集的结果,作者将分析限于经过实验测试的918种药物,将E918列表中临床试验中的37种强效药物视为阳性,而将其余881种药物视为阴性。由于这些试验的结果基本上是未知的,因此针对CT415数据集的验证测试了每套流程预测药物界对COVID-19患者具有预期潜在疗效的药理学共识的能力。
对于E918实验结果(图4A),曲线下的最佳面积(AUC)为P1的0.63,接着依次为P2(AUC = 0.58)和P3(AUC = 0.58)。对于CT415(图4B),观察到四个基于AI的流程预测能力最强(AUC为0.73-0.76),紧随其后的是P1(AUC = 0.57)和P2(AUC = 0.56)。
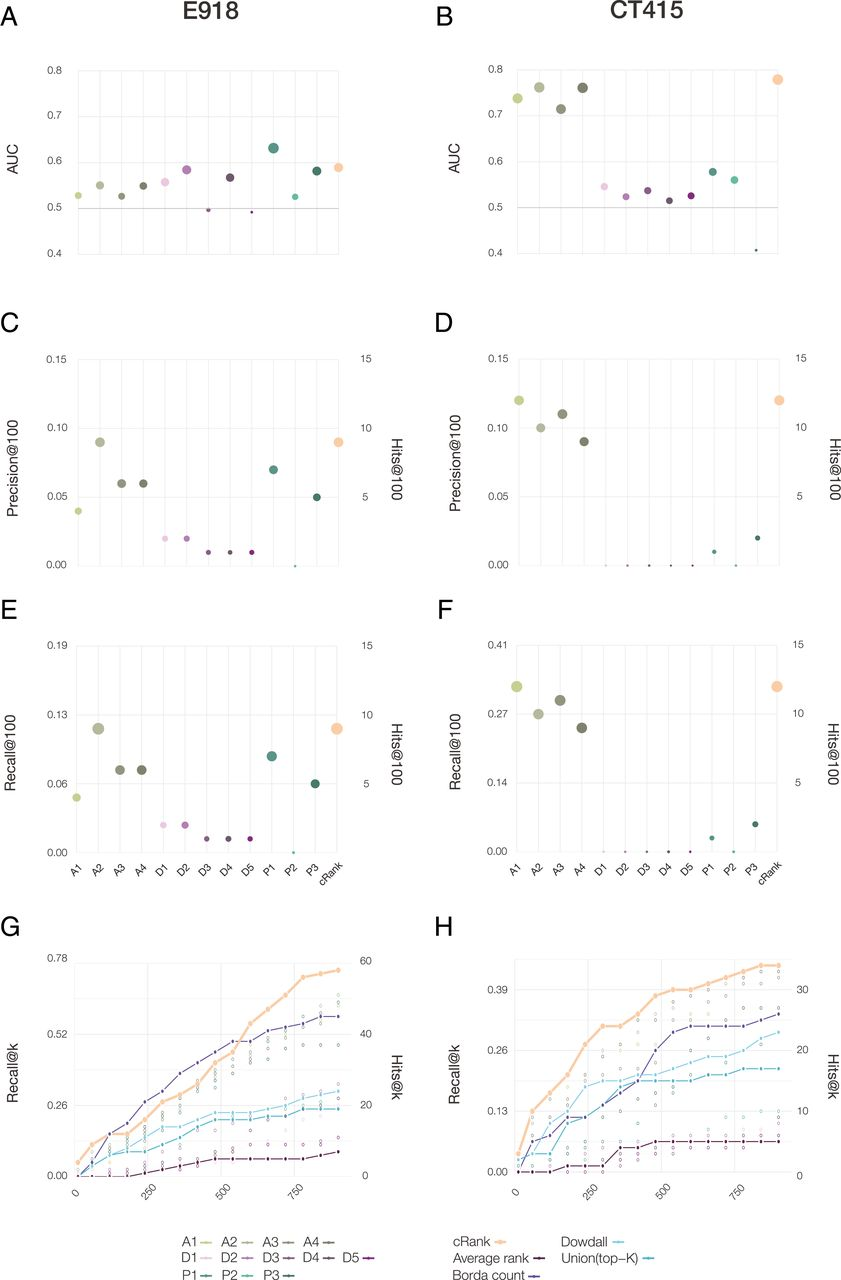
Fig. 4.
Performance of the predictive pipelines. (A and B) AUC, (C and D) precision at 100, and (E and F) recall at 100, for 12 pipelines tested for drug repurposing, each plot using as a gold standard the S&W drugs in E918 (left column) and drugs under clinical trials for treating COVID-19 as of April 15th, 2020 (CT415, right column). (G and H) The top K precision and recall for the different rank aggregation methods (connected points), compared to the individual pipelines (empty symbols) documenting the consistent predictive performance of CRank. Similar results are shown for two other datasets in SI Appendix, Fig. S8: the prospective expert curated E74 and the clinical trial data was refreshed on June 15, 2020 (CT615).
通过比较各个流程的结果,作者的第一个关键结果是发现,尽管大多数算法都显示出统计上显着的预测能力,但它们在不同的真实数据集上具有不同的性能:AI流程提供了对于临床试验选择的药物的强大的预测能力,而临近算法可为E918实验结果提供更好的预测能力。虽然这12套流程在前100名中识别出22种阳性药物,但没有一条流程能够为所有实验结果提供一致的优异性能,而这促使作者开发出一种多模式方法,可以提取所有流程的联合预测能力。
药物再利用的多模式方法
作者发现,等权重投票性能最差,落后于大多数独立流程的预测能力(图4G、H)。The Union List和Dowdall提供了更好的预测结果,但落后于最优的单个流程。Borda方法对于E918具有很强的预测性能,但对CT415则没有。相反,依靠贝叶斯因素的CRank为所有数据集和大多数K值(当实验验证能力有限的情况下,由预测流程给出K种药物,并使这K种药物种阳性药物尽可能多)提供了一致的高预测性能。CRank在其他两个数据集上的表现同样出色:手动编制的预期清单E74(在讨论中描述)和2020年6月15日更新的临床试验清单(C615)。
换句话说,作者发现CRank提取了所有方法的累积预测能力,匹配或超过了所有数据集中各个流程的预测能力,这是作者的第二个关键结果。这也表明,在没有基本事实的情况下,当各个流程都具有一定的预测能力时,寻求在各个流程之间成对分歧数量最少的排名的Kemeny共识(CRank是作者对于Kemeny共识是难以计算的NP问题的解决方案之一)代表了一种有效且理论上合理的策略。
网络效果
大多数以计算为依据的药物再利用方法都依赖于docking patterns,因此仅限于与病毒蛋白或病毒蛋白宿主靶标结合的化合物(图1C)。一个很好的例子是remdesivir,一个直接作用的抗病毒药,可以抑制病毒的RNA聚合酶。相反,作者的流程也可以识别靶向宿主蛋白的药物,以诱导基于网络的干扰,其中一些可能会改变病毒进入细胞或在细胞内复制的能力。在完整的宿主中,这些药物也可能通过其他机制起作用(例如地塞米松等皮质类固醇的抗炎作用),而这往往只能在动物模型或人体试验中评估。
我们发现,77种S&W药物中只有一种直接靶向病毒蛋白结合靶标:amitriptyline,其靶向SIGMAR1(NSP6 SARS-CoV-2蛋白的靶标)。换句话说,在作者实验筛选中显示功效的77种药物中,有76种是“网络药物”,它们通过扰动宿主亚细胞网络来达到其效果,这是我作者的第三个关键发现。确实,由于网络药物不靶向病毒蛋白或其宿主靶标,因此无法使用传统的基于结合的方法进行鉴定。但是,它们已通过基于网络的方法成功地确定了优先级。

 浙公网安备 33010602011771号
浙公网安备 33010602011771号