文献阅读 | Analysis of the recombination landscape of hexaploid bread wheat reveals genes controlling recombination and gene conversion frequency
Gardiner, LJ., Wingen, L.U., Bailey, P. et al. Analysis of the recombination landscape of hexaploid bread wheat reveals genes controlling recombination and gene conversion frequency. Genome Biol 20, 69 (2019). https://doi.org/10.1186/s13059-019-1675-6
该研究报道了交换和基因转换在六倍体小麦全基因组水平的分布规律,并克隆了一个影响交换和基因转换发生频率的基因RecQ,该基因是在小麦中发现的影响基因转换频率的首个基因,具有重要的育种价值。
Analysis of the CO landscape in wheat
为了研究面包小麦的重组 landscape ,作者使用了13个测定了基因型的重组自交系(RIL)群体,这些群体是利用英国优良品种 Paragon 的单种子后代与不同的地方品种和优良材料杂交而成的。通过使用父母的基因型,可以准确地绘制基因组中 crossover(CO) 的位置。除了大型的CO区块,基因型的较短位移在整个基因组中也经常遇到。它们以前被忽视为潜在的基因分型错误或与标记的局部排序问题。这些短暂的转移是基因转换(gene conversion, GC)事件的潜在标志。小麦基因组序列组装和重叠群的伪染色体(pseudo-chromosomes)局部排序的最新进展使作者能够更有把握地将小麦短位移归类为GC。
对于13个群体中的每个群体,每个重组自交系的 CO 数量呈现了相对正态分布(图1A),并在每个群体中都有发现一些离群值。在13个群体中,每个RIL的平均 CO 频率保持相对稳定,从40.8到51.9不等,与类似研究一致。
对于每个群体,作者计算了共享同一个 CO 位点的RIL的数量。图1b突出显示了共有 CO 位点的分布;平均而言,共有10.2%的 CO 位点的峰值仅出现在一个RIL中,其余的出现于两个或更多群体中。随着RIL数目的增加,CO保守率稳步下降。具有保守CO点的RIL最大数量从未超过种群规模的55.3%(范围44.6-55.3%)。虽然作者并不期望在这种规模的群体中有高度保守的CO位置,但是的COs的一部分重叠可能是由于作者将snp组合成了20mbp窗口来寻找CO导致的。
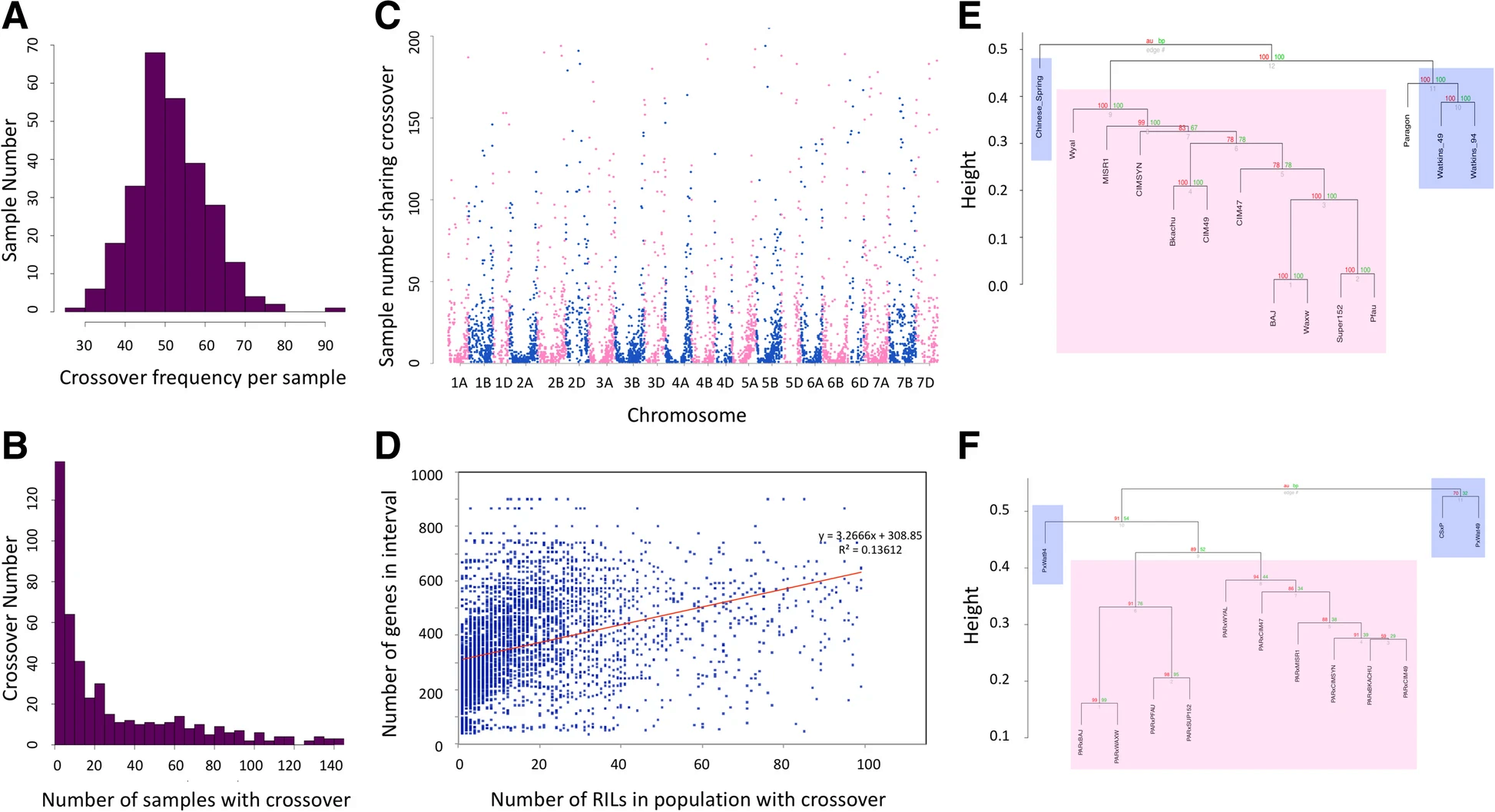
图1
小麦重组景观。a在Paragon×CS 群体中记录的每个RIL的COs数(每个样本的CO频率)频率直方图。b共享每个记录的CO的RIL数(每个CO的样本数)作为 Paragon×CS 种群的频率直方图。c对于所有分析的COs,CO的位置绘制在x轴上,y轴代表共享CO的群体内样本数量。d两个20 Mbp窗口的交集定义了一个CO。因此,对于包含中心定义CO的所有40 Mbp窗口,在每个区间内发现的高置信度基因的数量与每个群体内的RIL数量一起绘制。e13个群体的亲本根据35K SNP阵列中的代表性等位基因聚类。f13个种群根据各自的CO谱进行聚类,即种群中每个记录CO的RIL数。在e和f中的树状图是使用基于相关的相异矩阵的R包pvclust平均连锁法生成的,聚类之间的距离度量值表示为y轴上的高度。AU(近似无偏)p值通过多尺度bootstrap重采样(bootstrap数为1000)计算。地方品种用蓝色方框突出显示,纯育种系用粉色方框突出显示
观察到, CO 位点集中在染色体末端,如果 CO 位点更频繁地出现在多个 RILs 中,这种影响就更加明显。CO 位点对基因区域有偏倚,具有统计学意义(双尾 t 检验 p < 0.0001,t = 6.2534,df = 10,174)。在定义的含 CO 位点区域,即代表 cM bin的窗口中,平均基因数为363.83个,平均由基因代表的区域比例为2.65% ,而在所有 SNP 窗口中,每个区域的平均基因数为342.98个,由基因代表的每个区域的平均比例为2.54% 。另外,CO 位点基因数目与重组近等基因数目与 CO 呈显著正相关(图1d,Pearson 相关系数(r) = 0.271706,n = 4780,p < 0.00001)。因此,如果在更多的RIL中观察到 CO,那么它更有可能是基因相关的。
作者评估了观察到的每个确定的CO的RIL数量,发现在一个群体中更常见的CO更可能在所有13个群体中出现。当群体中有更多的RIL具有CO时,仅在一个群体中看到的COs数量减少;相反,当群体中有更多的RIL具有CO时,在所有13个群体中看到的COs数量增加。
GCs are more prevalent than COs in wheat
以前,我们通过比较代表 cM bin的窗口来定义 COs。这种方法不受小麦品种间基因组组织细微差异的影响,例如 GC (gene conversion) 事件。Sun 等人报道了拟南芥中每次减数分裂有120-222个 DSB(double-strand breaks,双链断裂),尽管所有 DSB 都有可能出现相关的 GC,但他们预测,假设错配修复使得50% 的 DSB 恢复到原来的等位基因状态,GC 数为60-111。由于 这些GC频率和GCs以前的近似值在2 bp和10 kbp之间,作者的分析可能会漏掉这些事件,因为每个群体平均有4335个 SNPs 可供分析。然而,在对COs的平行定义进行调整后,我们能够在13个群体中平均每个RIL确定 104个潜在GCs
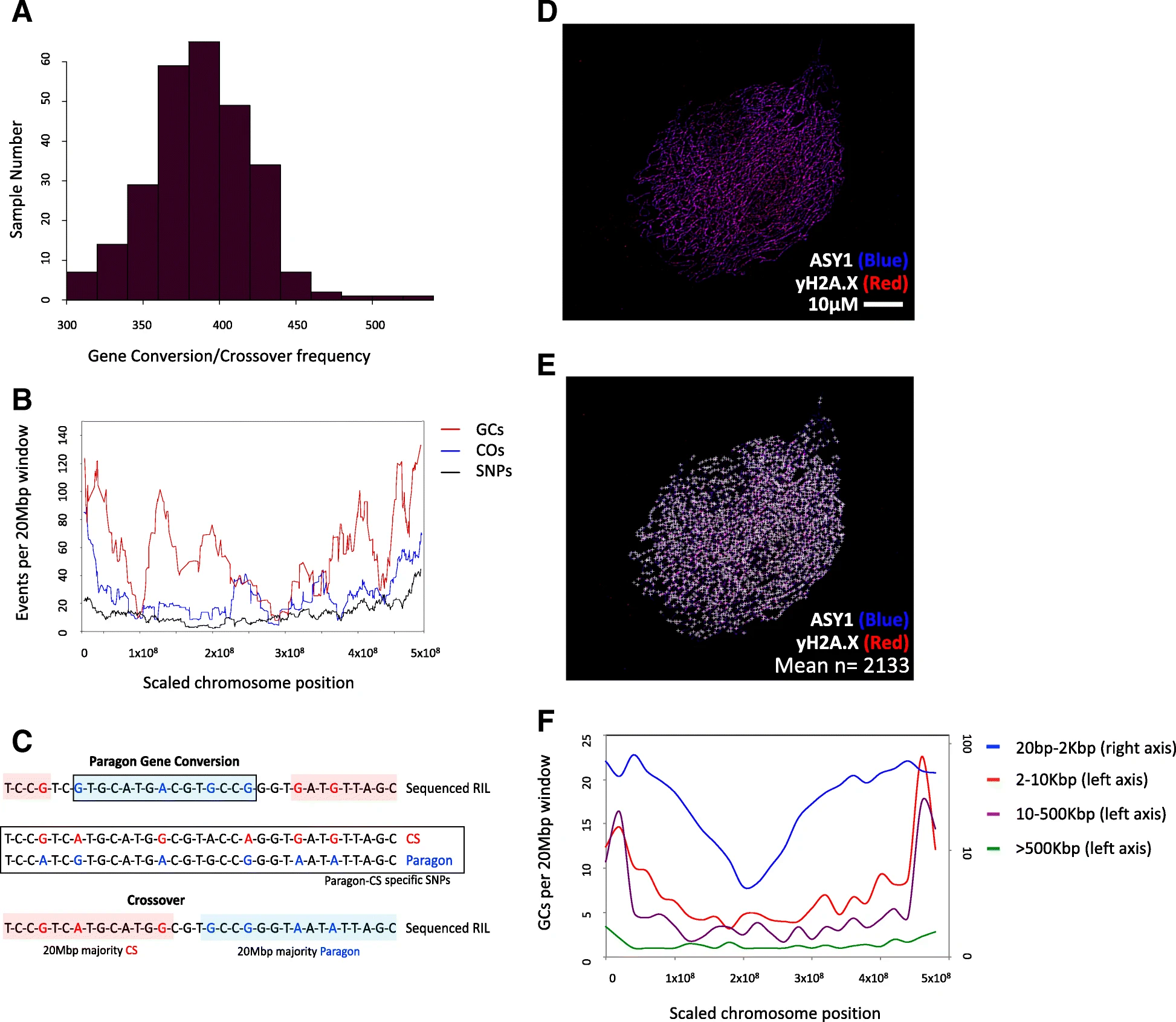
图2
序列交换事件的精细尺度分析。a 用频率直方图记录Paragon×CS群体中每个重组自交系的 COs 和/或 GC 数(每个样本 GC/CO 频率)。b 每个20-Mbp 窗口分别绘制 COs (COs)、 gc (gc)和 array SNPs 的数量折线图。所有染色体归一化为500mbp 长度,显示在一个单一的图中。显示了每个数据集的移动平均值(周期 = 15)。c 使用预先定义的Paragon和CS特异性纯合SNPs在skim测序数据中 call
基因转换(GCs)和交叉(COs)的方法示意图。d 染色体轴蛋白ASY1(蓝色)和yH2A.X(红色)的免疫定位六倍体小麦瘦素雄性减数分裂细胞核DNA DSB的标记。比例尺 = 10 μM。e 原始细胞核为 d,但 yH2A.X 与 ASY1同位化。显示的图像 n = 1673显示了5个复制区的 yH2A.X 焦点的平均数。f 在整个基因组中,每20 Mbp窗口长度为20 bp–2 kbp、2–10 kbp、10–500 kbp和>500 kbp的GC数分别绘制折线图。染色体按b标准化,显示每个窗口的平均频率
QTLs identified for CO frequency
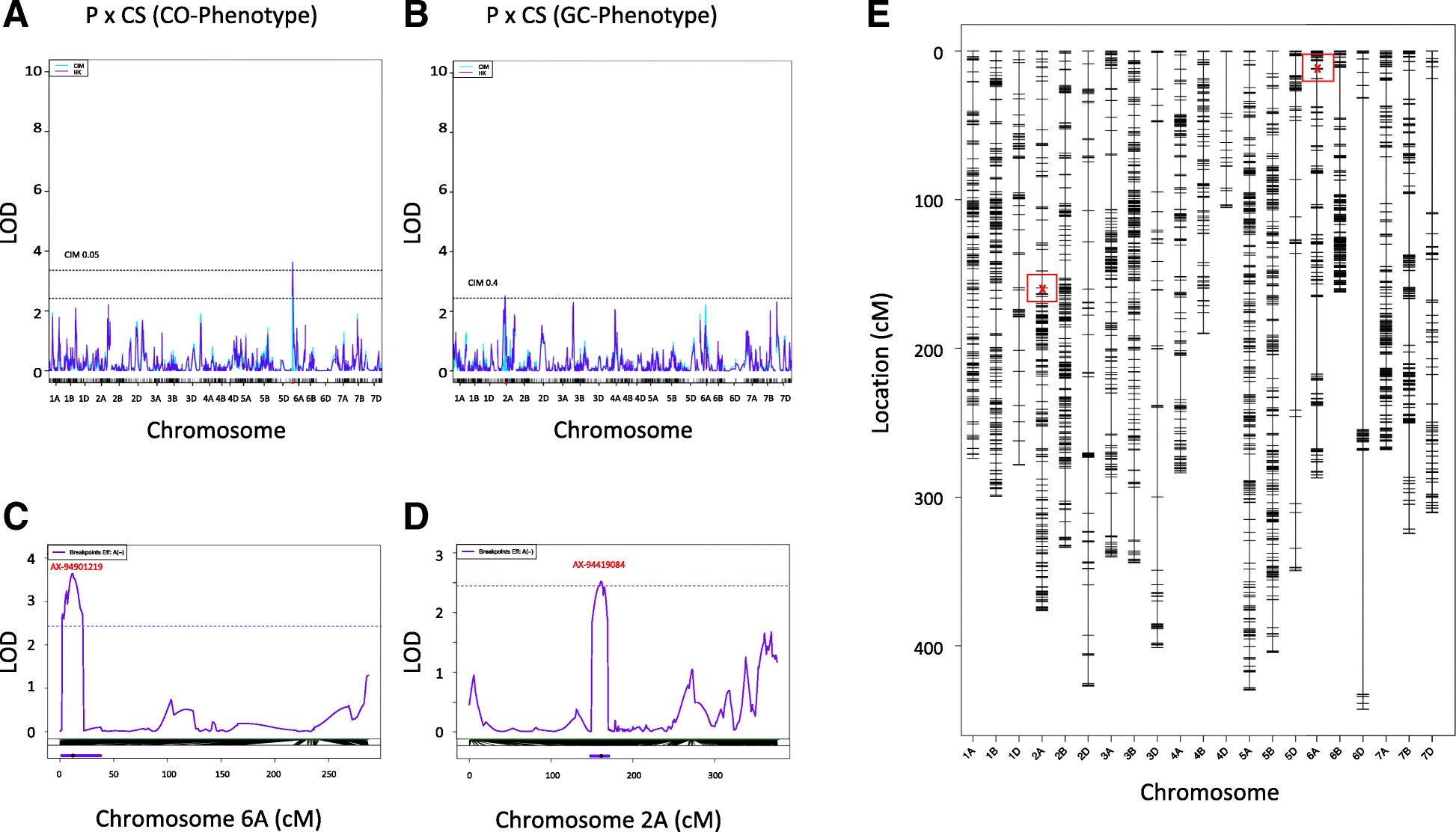
图3
利用 Paragon×CS 群体进行 QTL 分析。Paragon×CS群体的 QTL 分析结果表明,a co-表型和 b gc-表型之间存在显著相关(p < 0.05)。详细绘制了各连锁群(即染色体)的 LOD 分数。提高c co-表型和 d gc-表型 QTL 峰的分辨率。e最后,显示峰关联的array SNP的位置用红色标记,用红色框包围,同时还显示每个染色体的所有其他阵列SNP位置
对于 GC 频率特征 ,作者确定了多个稳健 QTL,它们解释了4.2-10.5% 的观测变异。作者只确定了13个群体中的4个群体的 QTL。
Validation of candidate genes

图4
从 RecQ-7和 RuvB 的 QTL 分析中检测候选基因。a 将敲除的RuvB系和对照系进行箱线图比较,根据CO-表型用折叠连锁窗定义共频率。b 基因敲除RecQ-7系和对照系的箱线图比较,用GC表型确定CO/GC频率。c 敲除RecQ-7系和对照系的箱线图比较,用共表型确定共频率。 d 多个物种(拟南芥、水稻和小麦)中与RecQ解旋酶家族序列相似的已鉴定基因的系统发育树,包括用于比较的小麦候选RecQ-7基因。Bootstrap values≥ 90%在分支上显示为绿点

 浙公网安备 33010602011771号
浙公网安备 33010602011771号