文献阅读 | Novel approach for parallelizing pairwise comparison problems as applied to detecting segments identical by decent in whole-genome data
Sapin, Keller (2021) Novel Approach for Parallelizing Pairwise Comparison Problems as Applied to Detecting Segments Identical By Decent in Whole-Genome Data. Bioinformatics . http://dx.doi.org/10.1093/bioinformatics/btab084
在基因组学研究中,数据集已经十分庞大,并且在未来数据规模仍会进一步增加。因此,需要成对比较的问题(无论是成对的SNP或是成对的个体)在计算上都将极具挑战性。对此,作者提出了一个解决两两比较的通用算法,以将 \(n^2\) 规模的问题简化为多个复杂度为 \(n\) 小问题并能够并行进行。
算法描述
本质上,作者的方法创新在于提出了一种近乎等大地划分子集的方法,从而将全部两两比较(\(n^2\)个)划分为等大的若干个子集进行运算,进而可以利用并行方法缩减时间。
首先,记所有构成一个个体的各项属性为 \(x_i, i \in \{1...n\} (n \geqslant 4)\),因此对于任意成对个体,可记为 \(\{x_i, x_j\}\)。记 \(p\) 为最小质数,则有 \(p^2 \geqslant n\)。为了创建子集,从 \(1\) 至 \(p^2\)的数字被按列放置在一个 \(p \times p\)的矩阵 \(P\) 中(图一)。其中,数字 \(1...n\) 是样本中个体的下标,数字 \(n+1...p^2\) 是未被关联的(are unassociated (null))而这将使一些子集拥有比其他子集更少的样本。

图1
Matrix \(P\), \(p_1\) and \(P_{p-1}\)
(第一行第一个矩阵为\(P\),第二个为\(p_1\),第三个为\(p_2\), 第二行第一个为\(p_3\),第二个为\(p_4\)。显然,\(p\)是\(5 \times 5\)的矩阵,因此\(p=5\)。)
如此一来,作者的方法即可产生 \(p^2 + p \approx n + \sqrt{n}\)个独立的子集。其中,第一个\(p\)子集由矩阵\(P\)的每一列定义,而下一个\(p\)子集由矩阵\(P\)的每一行定义,即共有\(2p\)个子集被从矩阵\(P\)中提取出来。举个例子,基于矩阵\(P\)的行定义的最后一个子集将包括个体\(x_p, x_{2p}, x_{3p}, ..., x_{p^2}\)。
剩余的 \(p^2 + p - 2p = p^2 - p\) 个子集,则由新矩阵\(P_k\)所定义。每个矩阵\(P_k (k \in \{1...p-1\})\)都是通过移动矩阵\(P\)的列中的元素得来的。与之前\(2p\)个从矩阵\(P\)中产生的子集不同,基于\(P_k\)产生的子集仅由\(P_k\)的行(而非列)所定义。因此,每个\(P_k\)矩阵贡献了\(p\)个额外的子集。为了生成这\(p-1\)个\(P_k\)矩阵,\(P_k\)中的每一列\(i\)中的每个元素都相对于\(P\)中的\(i\)被向上移动了\(r(i,k)\)行,并有:
\(r(i, k) = ((i - 1) \times k) \,\,\,\, mod \, p\)
其中,\(mod\)为取模。举个例子,对于\(k=1\),则有\(r(i=1, k=1)=0\),因此\(P_1\)的第一列不做移动与\(p\)的第一列保持一致;\(r(i=2, k=1)=1\),因此P_1\(的第二列中的元素相较于P\)中的上移了一个元素;\(r(i=3, k=1)=2\),因此P_1\(的第三列中的元素相较于P\)中的上移了两个元素,以此类推。
总之,产生的子集共有\(p^2+p\)个(其中,\(2p\)个是由原始矩阵\(P\)生成的,剩下的是由矩阵\(P_k\)生成的),每个个体都出现在了\(p+1\)个不同子集中,而每个子集中的个体数为\(p\)(或者少于\(p\),在\(p^2>n\)的情形下)。图2展示了当\(n=p^2=25\)时的矩阵\(P\)、\(P_1\)、\(P_2\)、\(P_3\)和\(P_4\)。
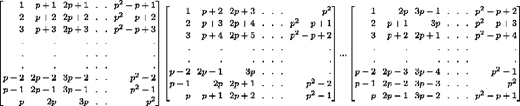
图2
Matrices \(P\), \(P_1\), \(P_2\), \(P_3\) and \(P_4\) for \(n=p^2=25\)
为了验证这一方法的准确性,作者计算了每一种样本对出现在同一子集中的次数\(t\)。经过对所有\(n<5M\)的模拟,作者发现每一种样本对均出现且仅出现在一个子集中。虽然没有严格的公式证明,但根据这一模拟结果,至少在\(n<5M\)的范围内,作者验证了其提出的方法是正确的,并且“没有理由怀疑该算法在\(n>5M\)时会表现得不一致”。因此,这些子集形成了一个仿射平面,其中子集是直线,子集中的个体是直线上的点。
不包括自我比较,根据作者提出的方法进行的比较总数是\(\frac{p^4-p^2}{2}\),是\(p^2+p\)子个集和每个子集内部\(\frac{p^2-p}{2}\)次比较的乘积。在naive方法下,整个数据集中的每一对个体都进行比较的话,总数是\(\frac{n^2-n}{2}\)。因此,在\(p^2=n\)的最佳情况下,作者提出的方法同native方法具有相同的运算次数,但可以进行并行化处理从而提升速度。类似地,如果包括自我比较,并仅对前\(p\)个子集执行这些比较以避免多次自我比较,再次导致与native方法相同的比较次数。GitHub页面位于https://github.com/emmanuelsapin,使用代码生成组成每个子集的个体ID列表。

Benchmark
假设对可以同时并行运行的任务数量没有限制,并且运算时间与比较次数成正比(1:1),作者提出的方法应该将成对比较问题所花费的时间减少n倍(即从\(n^2\)变成\(n\))。这是由于作者的方法使得每个子集内部进行了$\approx \frac{p^2}{2} \approx \frac{n}{2} \(次比较,而标准方法则是在全部样本中进行了\)\approx \frac{n^2}{2}$次比较。
接着,作者讨论了实际运行环境中,并不一定能达到缩减\(n\)倍的理想效果,因为并行运算的作业调度、IO限制等都会导致实际运行时间的增长。
为了量化作者的方法在实际问题上的表现,作者采用了UK Biobank data中的部分样本(从\(n=100\)到\(n=435187\),即整个数据集)进行IBD比较。比较采用了GERMLINE软件。同时,由于比较次数为\(n^2\)的传统方法需要的内存(约2.4TB)和时间(约1年)太大太长,作者只能以较小样本估计整个数据集两两比较时所需时间。
为了进行作者所需的并行运算,作者采用了University of Colorado at Boulder的运算集群the Blanca Condo Cluster。同时,作者对GERMLINE软件进行了“minor modification”以减少硬盘读写次数,并表示这一改动使得“文件读取操作的总数减少了三个数量级”。
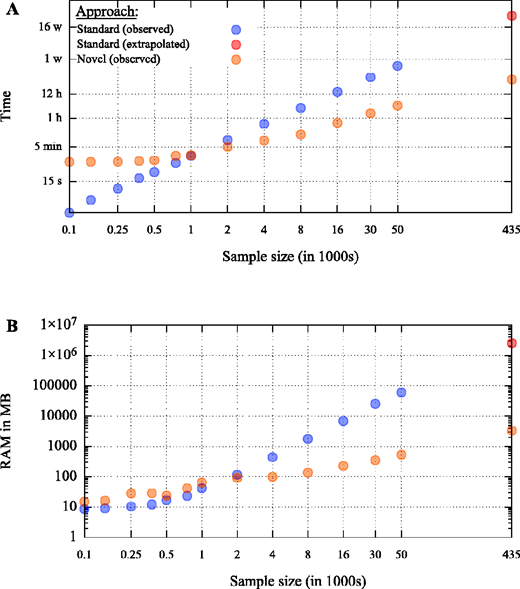
图3
两种方法用时(A)和内存占用(B)的比较。

 浙公网安备 33010602011771号
浙公网安备 33010602011771号