
ESM-1v | 语言模型赋能蛋白功能零样本预测(3)
Language models enable zero-shot prediction of the effects of mutations on protein function
1. Introduction
蛋白质具有多种多样的功能,这些功能构成了生命的复杂性。蛋白质序列通过自发地将序列折叠成蛋白质的三维结构来编码功能。序列突变对功能的影响形成了一个景观,揭示了功能如何约束序列。蛋白质序列中的某些位点的改变是不能被容忍的,因为它们对蛋白质的功能至关重要。其他位点是因为它们共同决定了结构和功能而演化的。突变可以增强蛋白质的活性,减弱它,或者保持不变。
序列变异对功能的影响可以通过深度突变扫描实验来测量。由数千到数十万的蛋白质功能测量组成,深度突变扫描为我们提供了关于蛋白质结构和功能的内在约束的见解。由于实施这种实验的成本和难度,深度突变扫描数据的汇编最多只包括对几十种蛋白质的实验,相对于人类基因组中编码的数万种蛋白质,以及我们希望了解的生命树上的数百万种蛋白质。
一个学习连接序列与功能的景观(landscape)的模型可以提供对功能的见解,而无需进行实验。可以从序列中学习无监督的突变效应模型。在一个家族的进化相关的蛋白质序列中的统计模式包含关于结构和功能的信息。这是因为蛋白质的属性作为约束通过进化选择序列。
在自然语言建模社区中,人们对零次迁移模型到新任务产生了兴趣。大型语言模型可以解决它们没有直接训练的任务。最近,蛋白质语言模型在各种结构预测任务中都达到了最先进的水平。迄今为止的工作主要集中在经典的表示学习设置中的迁移,使用预训练的特征并在下游任务上进行监督。
在这项工作中,我们展示了在大型和多样的蛋白质序列数据库上训练的语言模型可以预测蛋白质功能的实验测量,而无需进一步的监督。以前的工作主要集中在使用来自实验数据的监督来转移表示。我们发现语言模型可以在没有监督的情况下迁移到预测功能测量。语言模型对各种功能差异很大的蛋白质进行零次和少次预测突变效应。我们使用最先进的蛋白质语言模型 ESM-1b 和 MSA Transformer 进行实验。我们引入了一个新的蛋白质语言模型,ESM-1v,其零次性能与最先进的突变效应预测器相当。通过使用来自蛋白质家族的序列对模型进行微调,可以进一步提高性能。预测捕获了蛋白质的功能景观,与核心和表面的氨基酸保守模式相关,并确定了负责结合和活性的残基。
2. Zero-shot transfer 零次迁移
零次学习传统上描述的是将分类器扩展到在训练中未见过的一组新类。在自然语言处理中,这个想法已经被扩展到描述模型转移到完全新的任务而不需要进一步的训练。如 Larochelle et al.[18] Hugo Larochelle, Dumitru Erhan, and Yoshua Bengio. Zero-data learning of new tasks. In AAAI, volume 1, page 3, 2008. 所提出的零数据学习,这种关于迁移的观点已经成为最近理解大型语言模型泛化能力工作的核心。与表示学习的区别在于,模型直接使用,没有为任务提供额外的监督。这意味着任务必须纯粹从预训练中学习。
在这项工作中,我们对零次迁移的观点与 GPT-3 的描述相似,如 Brown et al.[27] Tom Henighan, Jared Kaplan, Mor Katz, Mark Chen, Christopher Hesse, Jacob Jackson, Heewoo Jun, Tom B. Brown, Prafulla Dhariwal, Scott Gray, Chris Hallacy, Benjamin Mann, Alec Radford, Aditya Ramesh, Nick Ryder, Daniel M. Ziegler, John Schulman, Dario Amodei, and Sam McCandlish. Scaling laws for autoregressive generative modeling. CoRR, abs/2010.14701, 2020. URL https://arxiv.org/abs/2010.14701. 所述。我们定义零次迁移是将模型转移到一个新任务,而不需要进一步的监督来专门化模型到任务。我们还考虑了与之密切相关的少次迁移的想法。在这里,如 Brown et al. 所述,我们定义少次设置是在推断时给模型作为输入的几个正面例子。与零次设置一样,没有进行梯度更新来专门化模型。与 Brown et al. 类似,这并不是一个超出分布的泛化的声明。假设是在预训练阶段,模型学习与它稍后将被转移的任务相关的信息。在蛋白质语言模型的情况下,预训练数据集包括来自整个进化过程的序列,这意味着模型可能会看到来自它将被评估的蛋白质家族的序列的例子。与计算生物学的标准方法的本质区别是,模型是通用的,可以在各种任务中应用,而不需要专门化。
对功能的测量是理解和设计蛋白质的中心重要性质,是研究蛋白质语言模型泛化能力的实际基础。深度突变扫描实验测量了数千到数十万种突变对单一蛋白质的影响,并已经在具有不同功能的各种蛋白质上进行了实验,使用了各种形式的实验测量。我们使用这些数据研究蛋白质语言模型对功能预测的零次和少次迁移。
无监督的突变效应预测器作为任务特定模型在来自单个蛋白质家族的序列上进行训练。在这种观点中,每个蛋白质都是一个独立的预测任务,目标是评估突变对蛋白质功能的影响。虽然在多序列比对(MSAs)上训练的突变效应预测器通常被描述为无监督的,但它们也可以被视为弱监督的。Hsu et al.[15] Chloe Hsu, Hunter Nisonoff, Clara Fannjiang, and Jennifer Listgarten. Combining evolutionary and assay-labelled data for protein fitness prediction. bioRxiv, page 2021.03.28.437402, mar 2021. doi: 10.1101/2021.03.28.437402. 观察到这样的模型通过 MSA 对任务有弱监督。
如果蛋白质语言模型可以从预训练中学习解决任务所需的信息,那么它们可以直接应用到任务的新实例上,而无需专门化。这意味着在实践中,可以训练一个通用模型,然后应用到各种可能的任务上。因此,零次和少次迁移代表了蛋白质语言模型可以为计算生物学工具箱带来的根本上新的无监督学习能力。
3. Method
使用mask语言建模目标训练的蛋白质语言模型被监督输出一个氨基酸在蛋白质的一个位置出现的概率,给定周围的上下文。我们使用这种能力来评分序列变异。对于给定的突变,我们可以考虑野生型蛋白质中的氨基酸作为一个参考状态,比较分配给突变氨基酸的概率与分配给野生型的概率。
我们使用在突变位置的对数几率比来评分突变,当在同一序列中存在多个突变 T 时,假设一个加性模型:
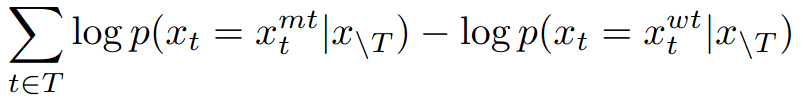
在这里,总和是在突变位置上,模型的序列输入在每个突变位置都被mask。
3.1 Zero-shot and few-shot transfer 零次和少次迁移
在零次设置中,直接对要评估的序列进行推断。由于 MSA Transformer 可以在推断时接受多个序列作为输入,我们在少次设置中使用这个模型,其中还提供了与要评估的序列一起的蛋白质家族的其他序列。在零次和少次设置中,只在推断期间执行模型的前向传递;不进行梯度更新。
3.2 Inference efficiency推断效率
与当前的最先进方法相比,使用 ESM-1v 进行推断更为高效。这是两个重要差异的结果:(i) 可以直接推断突变的效果,而无需训练任务特定的模型;(ii) 可以使用单次前向传递预测适应性景观。时间要求在图 7 中进行了总结。
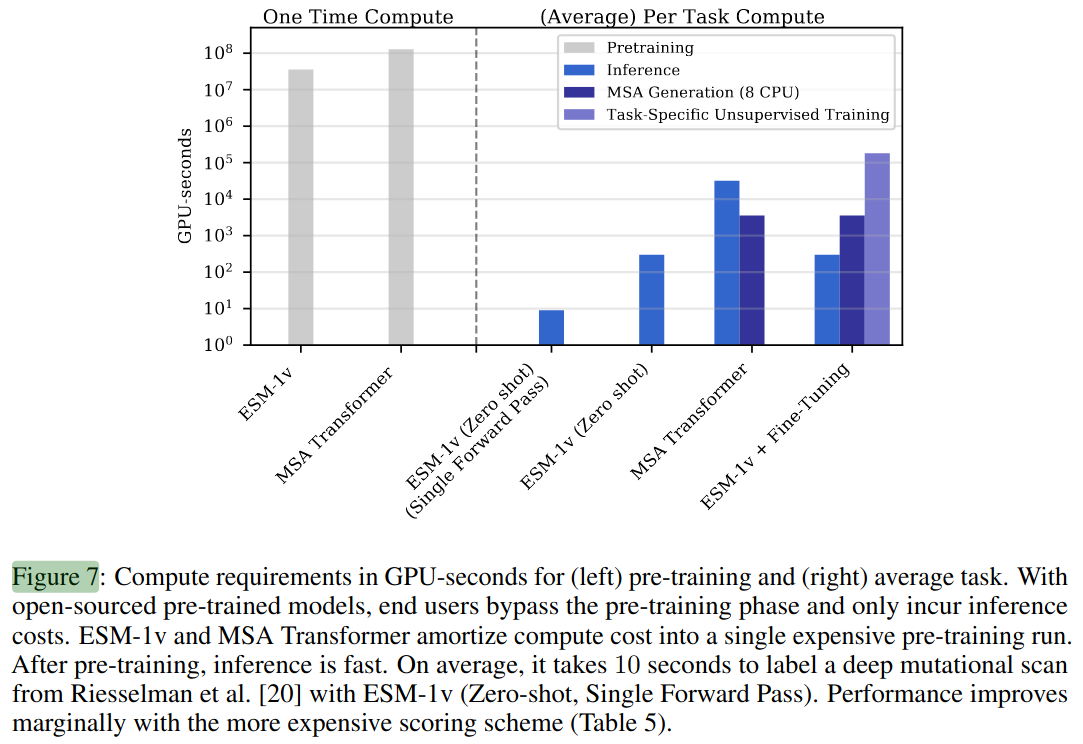
(左图)预训练与(右图)平均任务所需的GPU计算时间(以秒计)。若使用开源预训练模型,终端用户可跳过预训练阶段,仅需承担推理成本。ESM-1v和MSA Transformer通过单次高成本的预训练分摊计算开销,预训练完成后推理速度极快。以Riesselman等[20]的深度突变扫描数据为例,ESM-1v(零样本、单次前向传播)平均仅需10秒即可完成标注。若采用计算成本更高的评分方案,性能仅有小幅提升(见表5)。
3.3 Scoring with MSA Transformer使用 MSA Transformer 进行评分
我们使用 MSA Transformer 和上述方程中的对数几率比和加性模型对突变进行评分。然而,由于 MSA Transformer 使用一组序列进行推断,我们将要评估的序列作为第一个序列输入,并提供来自 MSA 的其他序列作为上下文。只对第一个序列进行遮罩和评分。
4 Results结果
4.1 Experimental setup实验设置
Prediction Models预测模型
我们与最先进的无监督变体预测方法,如 EV-Mutation 和 DeepSequence 进行比较。我们还检查了文献中最近介绍的各种蛋白质语言模型的性能。位置特定评分矩阵(PSSM)、EVmutation 和 DeepSequence 方法都是基于 MSA 的。PSSM 独立地处理序列中的每个位置,将似然因子化为每个序列位置的一个术语。EVmutation 是一个Potts模型,它增加了成对的术语,模拟位置之间的相互作用。DeepSequence 引入了一个潜在代码,允许位置之间的潜在高阶相互作用。UniRep、TAPE、ProtBERT-BFD、ESM-1b 和这里介绍的 ESM-1v,都是在大型未对齐和不相关的蛋白质序列数据库(例如 Pfam 或 UniRef)上训练的单序列语言模型。除了 UniRep,它使用下一个令牌预测进行训练,所有模型都使用遮罩语言建模进行训练。最后,MSA Transformer 是两种方法的结合;它在一个大型的 MSA 数据库上使用遮罩语言建模进行训练,并在推断期间将 MSA 作为输入。
ESM-1v
我们训练 ESM-1v,一个有6.5亿参数的变压器语言模型,用于预测变体效应,该模型在整个进化过程中的9800万个多样性蛋白质序列上进行训练。该模型仅在序列上进行训练,没有任何来自功能实验测量的监督。我们使用 Uniref90 2020-03,采用 Rives et al. 的 ESM-1b 架构和遮罩语言建模方法。我们训练五个具有不同种子的模型来产生一个集成。
Evaluation评估
模型在 Riesselman et al. 收集的41个深度突变扫描集上进行评估,这些扫描集包括评估一组多样性蛋白质的各种任务。在任务之间,实验在测试的功能和执行的测量中有所不同。我们将每个深度突变扫描数据集视为一个单独的预测任务,使用模型为数据集中的每个变体进行评分。任务被分为一个由十个突变扫描数据集组成的验证集和一个由其余数据集组成的测试集。我们通过使用Spearman等级相关性比较分数与实验测量来评估性能。
Comparisons比较
由于 EVMutation 和 DeepSequence 的已发布版本使用从 Uniref100 的早期版本生成的 MSAs,我们使用 EVMutation 方法和与我们的预训练数据集同时的 Uniref100 版本生成新的 MSAs。我们使用他们的开源代码训练 EVMutation 和 DeepSequence 的复制品。相同的 MSAs 也用于与 MSA Transformer 的少次实验和与 ESM-1v 的无监督微调实验。
4.2 Language models enable zero-shot and few-shot prediction of the effects of mutations语言模型使零次和少次预测突变效应成为可能
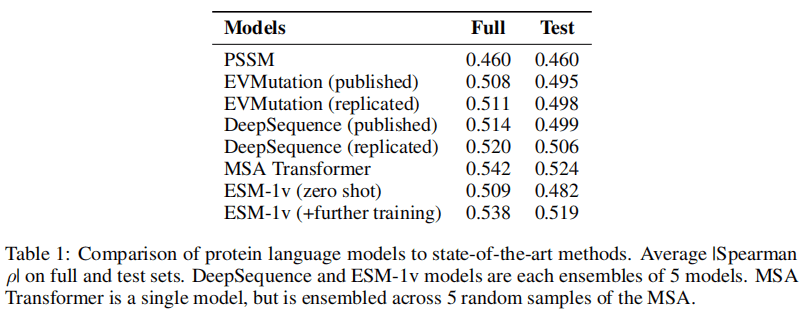
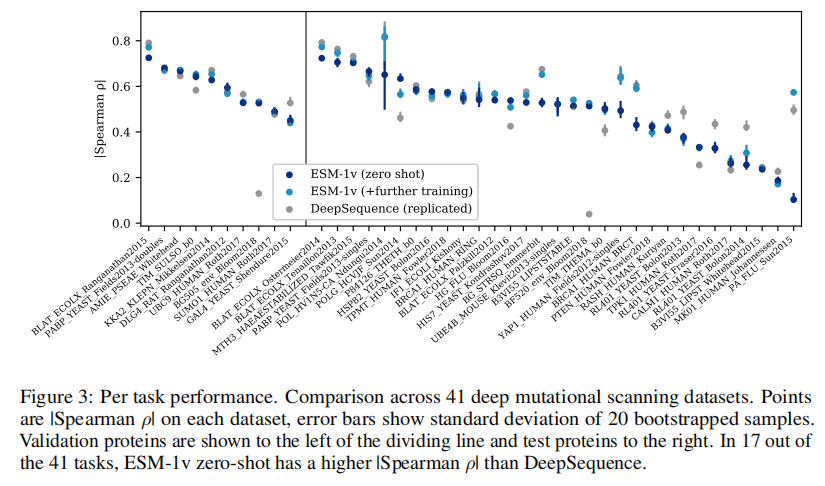
ESM-1v 和 MSA Transformer 模型做出了最先进的预测。表1比较了模型在41个突变扫描数据集上的整体性能。图3展示了 ESM-1v 和 DeepSequence 在每个任务上的比较。与 DeepSequence 相比,ESM-1v 的零次推断在41个数据集中的17个上与实验测量有更好的相关性。通过配对t检验,这两种方法在统计上是不可区分的。表2比较了零次设置中的蛋白质语言模型。ESM-1v 的性能超过了现有的蛋白质语言模型 TAPE、UniRep、ProtBERT-BFD 和 ESM-1b。
Pre-training data预训练数据
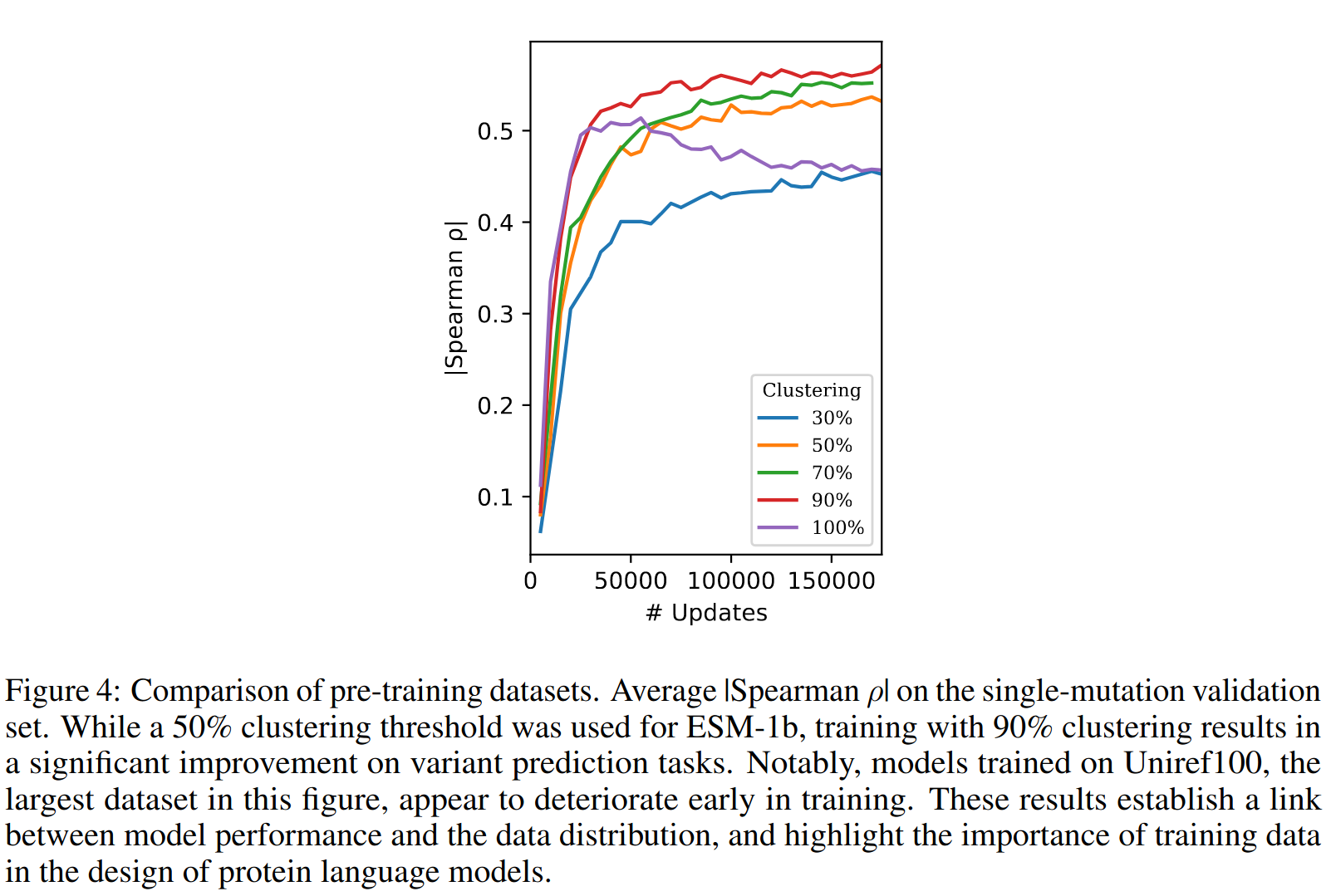
我们检查预训练数据的聚类级别对结果的影响。图4比较了在不同序列相似性阈值下聚类的数据集上预训练的模型。ESM-1b 是在50%的身份阈值上聚类的序列上进行训练的。使用70%的阈值可以看到改进,而在90%处看到最大的改进。尽管 Uniref100 是数据集中最大的,但在训练初期其性能似乎已经恶化。这些结果建立了模型性能与数据分布之间的联系,突显了训练数据在蛋白质语言模型设计中的重要性。
Scoring methods评分方法
我们在验证集上比较了四种评分方法 - 遮罩边际、野生型边际、突变边际和伪似然。方程 (1) 中描述的遮罩边际方法的性能优于其他评分方法,包括考虑非突变位置的似然变化的方法。评分方法在附录A中详细描述。
Parameter count参数计数
与蛋白质语言模型的先前工作已经建立了模型规模与蛋白质结构学习之间的联系。我们检查零次迁移性能作为参数计数的函数。我们使用与 Henighan et al. 中描述的相同的宽度、深度和学习率训练模型,观察到随着规模的增加而改进。这些发现表明,继续扩大模型规模将进一步改善结果。
4.3 MSA Transformer
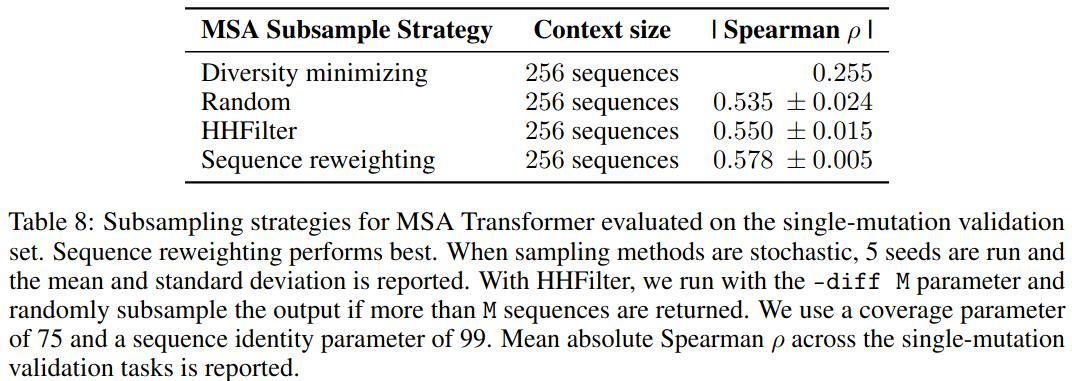
我们检查提供给 MSA Transformer 的序列如何影响少次迁移。表8比较了不同的序列选择方法,这些方法改变了序列的多样性。提供更多样化的序列集可以提高少次性能。选择一组最大化多样性的序列的性能优于选择最小化多样性的序列集。随机抽样表现得更好,根据序列权重抽样序列表现得最好。
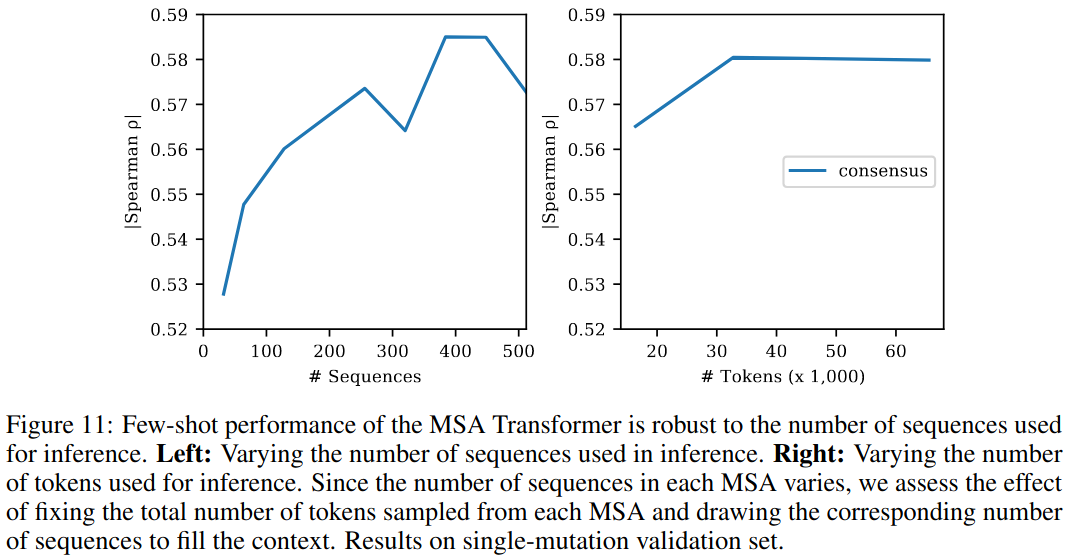
我们还改变了用于推断的序列数量。图11显示了作为输入给定的序列数量的函数的少次性能。该模型只使用少量序列就表现得很好,但使用总共384个序列时表现得最好。在主要的表格中,我们报告了使用序列重新加权和集成预测从 MSA 中的五个不同子样本中抽样的384个序列的结果。
4.4 Unsupervised fine-tuning on MSAs在 MSAs 上的无监督微调
虽然 ESM-1v 在零次设置中的评估表现良好,但我们探讨了是否可以通过在 MSA 上进行微调来改进结果。在之前的工作中,已经使用 MSAs 上的微调作为迁移学习的一个阶段,以专门化一个预训练的模型到一个蛋白质家族,然后再使用带标签的数据进行监督。在这里,我们考虑使用微调后的模型直接进行无监督预测,而不添加来自实验数据的监督。
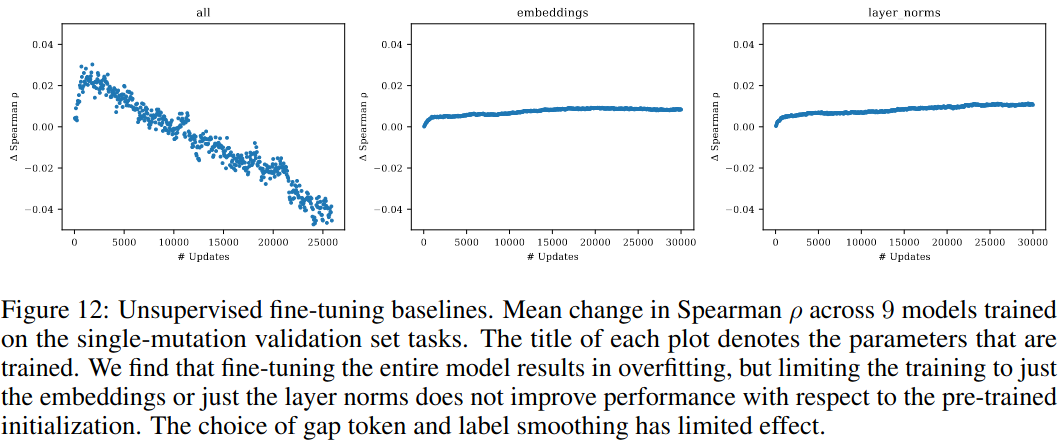
我们观察到,简单地在 MSA 上微调模型会导致快速过拟合和预测任务的性能差(图12)。虽然我们尝试了在微调过程中冻结参数的各种方法,详见附录B,但没有产生显著的改进。我们发现,使用预训练序列对微调进行正则化的方法表现得很好,并且可以训练所有参数而不会过拟合。尖峰微调将整个数据集的平均绝对Spearman rho从零次评估的0.510提高到微调后的0.537。
5 Analysis of models模型分析
Protein structure and function蛋白质结构和功能
ESM-1v 的概率反映了蛋白质内部位点的功能属性。我们使用模型对位置的预测的熵作为其对保守性的估计的度量。最低的熵预测集中在结合位点。图14比较了模型的熵在结合位点和非结合位点之间的分布。观察到结合和非结合位点残基的熵分配之间存在显著差异。图5可视化了模型在与其DNA底物相互作用的DNA甲基转移酶 M.HaeIII 的晶体结构上预测的10个最低熵残基的侧链。在晶体结构中,底物的胞嘧啶插入到酶的活性位点中。低熵残基集中在活性位点并与胞嘧啶相互作用。
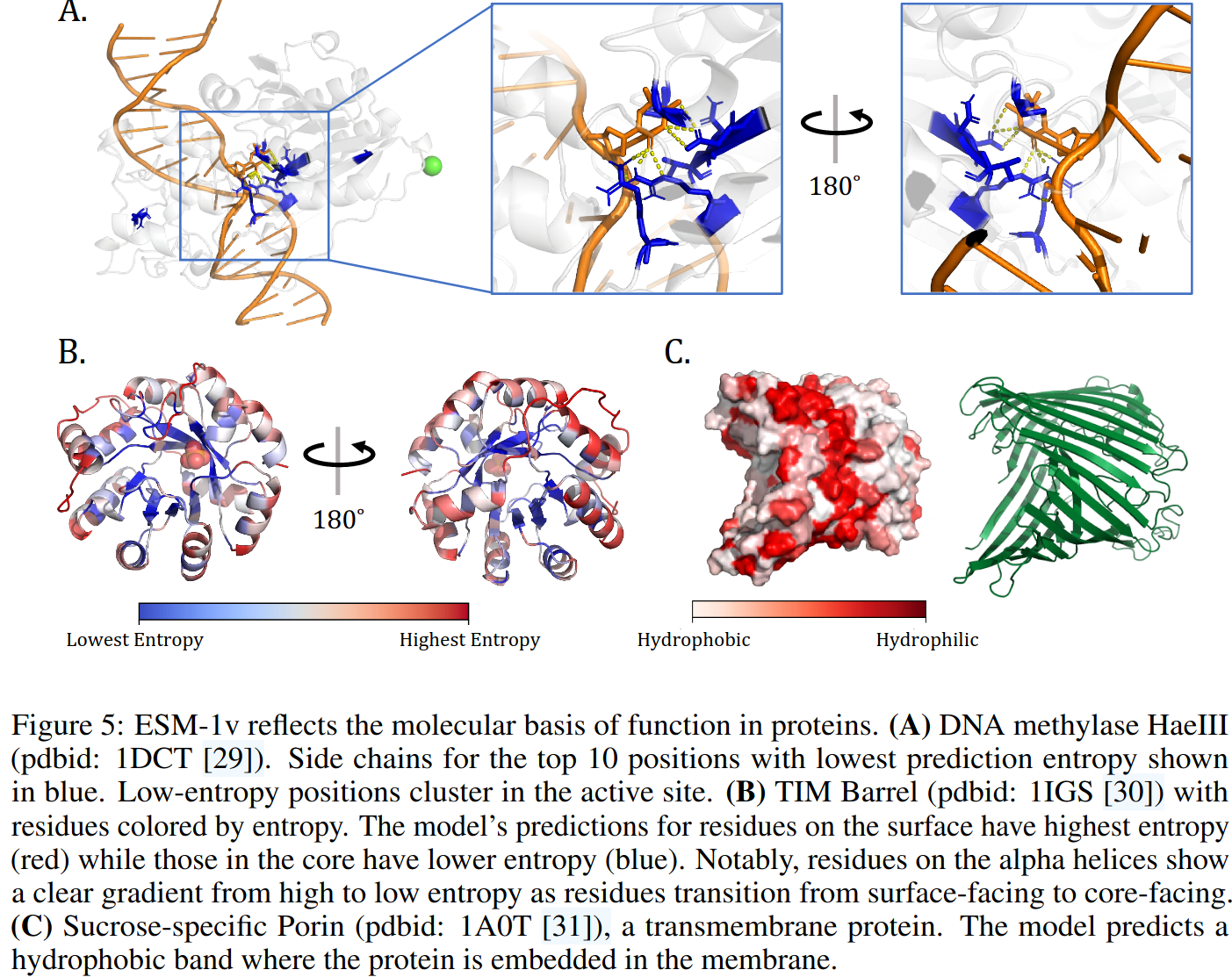
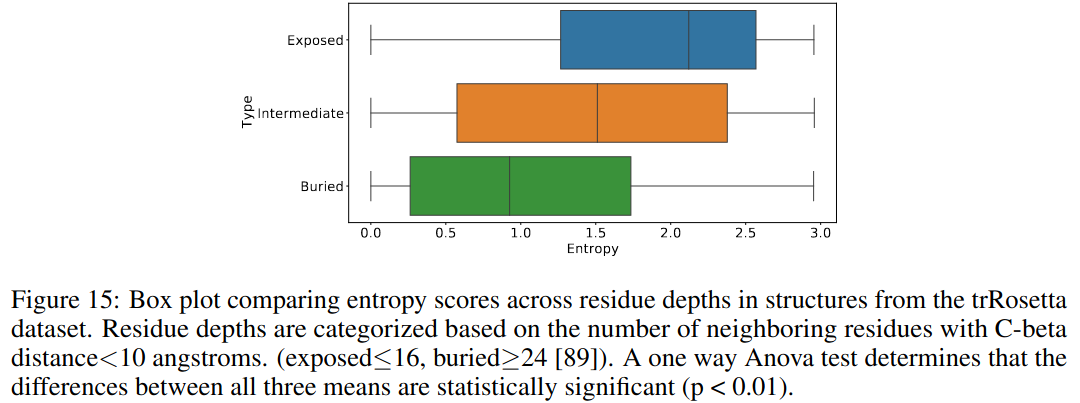
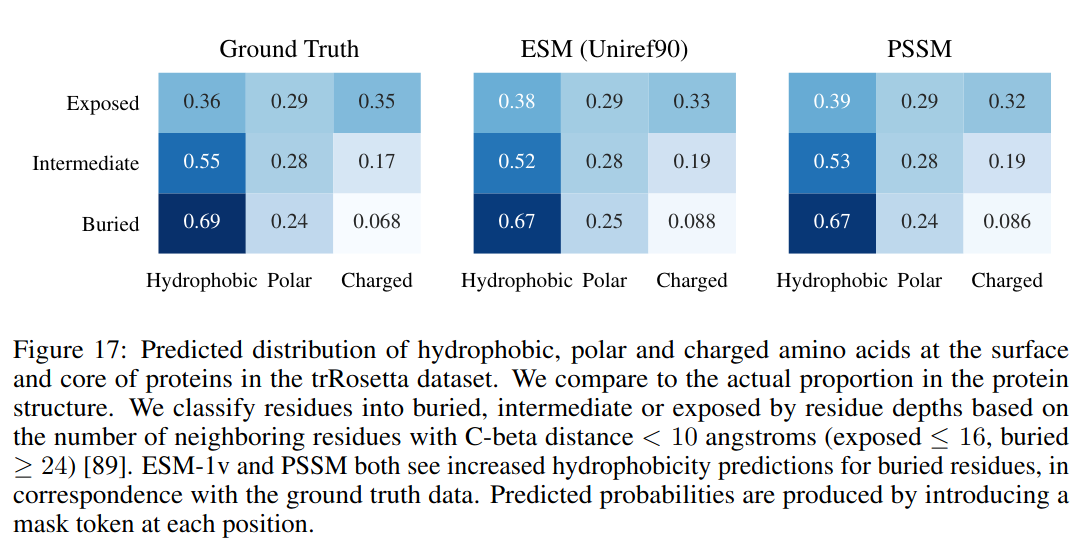
模型概率也对应于结构。图15比较了分配给蛋白质核心中的位点与暴露在表面的位点的熵。模型为蛋白质核心中的位点分配了显著较低的熵,这与核心中的紧密堆叠对残基选择施加更大约束的观点一致。图5B可视化了模型分配给每个位置的熵,叠加在 Indole-3-glycerolphosphate Synthase 的结构上,这是一个 TIM 桶蛋白。较高的熵分配给α螺旋上有向外的侧链的残基,而较低的熵分配给向内的位置。图17比较了对埋藏位点与非埋藏位点分配的疏水性、极性和带电氨基酸的概率。模型在核心中更喜欢疏水性残基,在表面上更喜欢亲水性残基。模型概率与经验概率和 PSSM 中的概率密切匹配。图5C可视化了模型在 Sucrose-specific Porin 的结构上分配给疏水氨基酸的概率,这是一个跨膜蛋白。模型在中心预测了一个疏水带,蛋白质在该带中嵌入膜中。
Calibration校准
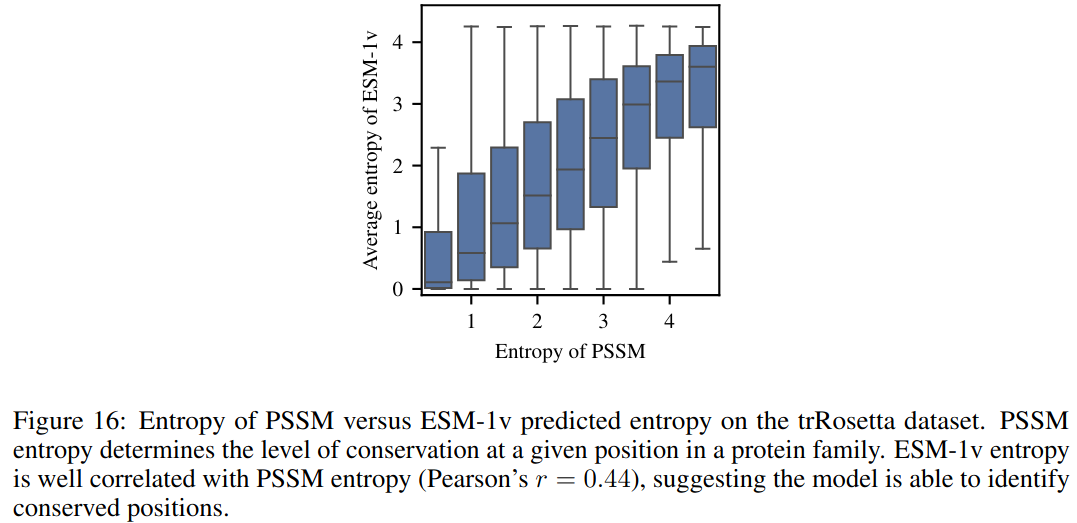
我们使用来自 trRosetta数据集的15008个长度小于1024的序列评估模型的校准。使用方程 (1) 中的遮罩边际概率计算每个位置的每个氨基酸的 ESM-1v 概率。图6显示,除了蛋氨酸外,该模型对所有氨基酸都进行了很好的校准。ESM-1v 总是预测蛋氨酸作为序列的第一个位置,因为完整的蛋白质序列总是从它开始,所以在将模型应用到子序列时必须小心。除了第一个残基外,模型达到了平均校准误差为0.006。我们还探讨了保守性(PSSM 的熵)与模型预测的熵之间的关系。图16显示,这两者之间的关系很好(皮尔逊的r = 0.44),这表明模型能够识别保守的位置。
6 Related Work相关工作
6.1 Protein language models蛋白质语言模型
在过去的几年中,许多团队已经为蛋白质序列开发了语言模型[21, 33, 14, 34, 35, 22, 13, 12]。这些模型已经被用于许多任务,包括有监督的低-N功能预测[16, 12],远程同源性检测[22, 12]和蛋白质生成[35]。通常的方法是迁移学习,其中一个预训练的语言模型被微调用于特定的问题。Vig等人[36]和Rao等人[26]发现,变压器注意力对应于已知的生物属性,如结构和结合位点,并可以用来预测接触。
6.2 Mutation effect prediction突变效应预测
已经开发了有监督和无监督的方法来预测突变效应。有监督的方法使用实验测量或来自临床变异数据库的标签来训练模型。可以使用包括线性回归、随机森林和支持向量机在内的标准机器学习工具[37]。已经为蛋白质专门设计了模型,使用特征工程,如 Envision [38] 和 PolyPhen-2 [39],集成方法如 Revel [40]、MPC [41]、CADD [42] 和 M-CAP [43],语言模型如 UniRep [21, 16] 和 ESM [12],以及其他表示学习方法[44, 45]。
无监督的突变效应预测器通过从原始蛋白质的进化景观中推断突变的可能性来工作。用于评分的是拟合到相关序列的密度模型。SIFT [46] 是使用位置特定评分矩阵的一阶方法。EVMutation [4] 通过在 MSA 上训练 Potts 模型将其扩展到二阶方法。DeepSequence [20] 通过在 MSA 上训练 VAE 来包括更高阶的交互,使用 ELBO 来评分突变。Riesselman等人[47] 提议使用不需要对齐序列的自回归模型。
Hsu等人[15] 显示,无监督的突变效应预测器可以扩展为执行有监督的预测,更好的无监督预测器通常会导致更好的有监督预测器。这表明改进无监督预测可以在两种设置中推动进展。与我们的工作同时,Hie等人[48] 使用开源的蛋白质语言模型 ESM-1b 和 TAPE 来预测蛋白质适应性景观中的进化方向。
7 Discussion
大规模语言建模的进步正使得蛋白质通用模型的目标更接近实现。这一研究方向希望能够建立一个模型,学会用其原生语言读写生物学,可以直接应用于一系列的蛋白质理解和设计任务。为了可扩展性,从序列中学习是很重要的:虽然没有高通量功能测量的中心数据库,且很少有编译存在,但在序列数据库中有数十亿的序列可以学习[49, 50]。序列为我们提供了一个无与伦比的视角,可以看到自然界通过数十亿年的进化创造出的分子部件的巨大多样性和复杂性。
无监督结构[51-53, 28, 54, 55]和功能[3, 4]学习方法首先有效地实现了这样一个想法:生物学特性可以直接从序列中读取,而不需要实验测量的监督。然而,这些方法并不是通用的,因为每一个要进行预测的蛋白质都必须训练一个专门的模型。我们展示了一个在许多不同的蛋白质家族上进行训练的通用模型可以实现相同的性能。与大型语言模型中关于三级蛋白质结构学习的观察类似[12, 26],我们发现增加模型的规模会提高功能学习的效果。模型中的突变景观的理解与蛋白质的功能的分子基础相关,捕获由折叠结构决定的结合位点和氨基酸偏好。
零次迁移是大规模语言模型的一个有趣的能力,并代表了与当前用于推断蛋白质结构和功能的无监督学习方法的基础相比的一个主要的转折点。零次迁移的能力意味着一个模型可以被训练一次,然后应用于执行许多任务的推断。它也是一个窗口,深入探讨从序列中学习可能的泛化形式。从序列中读取结构和功能设计原则是编写新的生物活性序列的必要能力。零次迁移设置中的泛化表明大型语言模型有潜力捕获可以转移到生成新的功能蛋白质的知识。

 浙公网安备 33010602011771号
浙公网安备 33010602011771号