R_Studio(聚类)针对iris数据比较几种聚类方法优劣
聚类分析 百度百科:传送门
聚类分析指将物理或抽象对象的集合分组为由类似的对象组成的多个类的分析过程
聚类与分类的不同在于,聚类所要求划分的类是未知的
聚类分析是一种探索性的分析,在分类的过程中,人们不必事先给出一个分类的标准,聚类分析能够从样本数据出发,自动进行分类。聚类分析所使用方法的不同,常常会得到不同的结论。不同研究者对于同一组数据进行聚类分析,所得到的聚类数未必一致。
IRIS (IRIS数据集) 百度百科:传送门
Iris数据集是常用的分类实验数据集,由Fisher, 1936收集整理。Iris也称鸢尾花卉数据集,是一类多重变量分析的数据集。数据集包含150个数据集,分为3类,每类50个数据,每个数据包含4个属性。
可通过花萼长度,花萼宽度,花瓣长度,花瓣宽度4个属性预测鸢尾花卉属于(Setosa,Versicolour,Virginica)三个种类中的哪一类。
一、k-means聚类


K-means聚类也称为快速聚类,k-means聚类涉及两个主要方面的问题。:第一,如何测试样本的“亲疏程度”;第二,如何进行聚类。通常,“亲疏程度”的测度有两个角度:第一,数据间的相似程度;第二,数据间的差异程度。衡量相似程度一般可采用简单相关系数或等级相关系数,差异程度一般通过某种距离来测度。k-means聚类方法采用第二个测度角度。k-means聚类的基本思想是先将样本空间分割成随意的若干类,然后计算所有样本点到各类中的距离,由于初始聚类结果是在空间随意分割的基础上产生的,因此无法确保所给出的聚类解满足上述要求,所以要经过多次反复。聚类数目确定本身并不简单,太大或太小都会失去聚类的意义。由于距离是k-means聚类的基础,因此也要注意:1、当聚类变量值有数量级上的差异时,一般通过标准化处理消除变量的数量级差异。2、聚类变量之间不应该有较强的线性相关关系。
iris2 <- iris iris2$Species <- NULL kmeans.result <- kmeans(iris2, 3) #将聚类结果与species进行比较 table(iris$Species,kmeans.result$cluster) plot(iris2[c("Sepal.Length", "Sepal.Width")], col = kmeans.result$cluster) points(kmeans.result$centers[,c("Sepal.Length", "Sepal.Width")], col = 1:3, pch = 8, cex=2)
数据预处理:从iris数据集中移除species属性
> iris2 <- iris > iris2$Species <- NULL > kmeans.result <- kmeans(iris2, 3) > #将聚类结果与species进行比较 > table(iris$Species,kmeans.result$cluster) 1 2 3 setosa 0 0 50 versicolor 2 48 0 virginica 36 14 0
绘制所有簇以及簇中心,值得注意的是多次运行得到的k-means聚类结果可能不同,因为初始的簇中心是随机选择的
plot(iris2[c("Sepal.Length", "Sepal.Width")], col = kmeans.result$cluster)
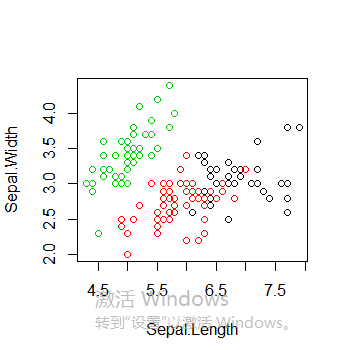
points(kmeans.result$centers[,c("Sepal.Length", "Sepal.Width")], col = 1:3, pch = 8, cex=2)

二、k-medoids聚类
k-medoids聚类 又被称为K-中心点聚类。函数pam()和pamk()都可以进行k-medoids聚类,k-medoids聚类与k-means聚类类似,主要区别是k-means聚类选择簇中心表示聚类簇,而k-medoids聚类选择靠近簇中心的对象表示聚类簇,在含有离群点的情况下,k-medoids聚类的鲁棒性更好,不像k-means聚类容易受极值影响。
library(fpc) iris2 <- iris iris2$Species <- NULL pamk.result <- pamk(iris2) pamk.result$nc #簇个数 table(pamk.result$pamobject$clustering, iris$Species) plot(iris2[c("Sepal.Length", "Sepal.Width")], col = pamk.result$pamobject$clustering) points(pamk.result$pamobject$medoids[,c("Sepal.Length", "Sepal.Width")], col = 1:3, pch = 8, cex=2) layout(matrix(c(1,2),1,2)) # 每页两张图 plot(pamk.result$pamobject) layout(matrix(1)) #改回每页一张图 library(cluster) pam.result <- pam(iris2, 3) table(pam.result$clustering, iris$Species) layout(matrix(c(1,2),1,2)) # 每页两张图 plot(pam.result) layout(matrix(1)) #改回每页一张图
数据预处理:从iris数据集中移除species属性
> library(fpc) > iris2 <- iris > iris2$Species <- NULL > pamk.result <- pamk(iris2) > pamk.result$nc #簇个数 [1] 2 > table(pamk.result$pamobject$clustering, iris$Species) setosa versicolor virginica 1 50 1 0 2 0 49 50
绘制所有簇以及簇中心
plot(iris2[c("Sepal.Length", "Sepal.Width")], col = pamk.result$pamobject$clustering)
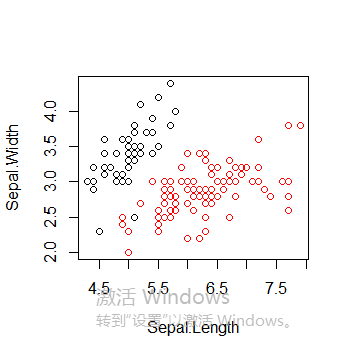
points(pamk.result$pamobject$medoids[,c("Sepal.Length", "Sepal.Width")], col = 1:3, pch = 8, cex=2)
图中可以看到聚类分为两簇,这是因为pamk()函数会调用pam()函数或clara()函数根据最优平均阴影官渡估计的聚类簇个数来划分数据
layout(matrix(c(1,2),1,2)) # 每页两张图 plot(pamk.result$pamobject) layout(matrix(1)) #改回每页一张图
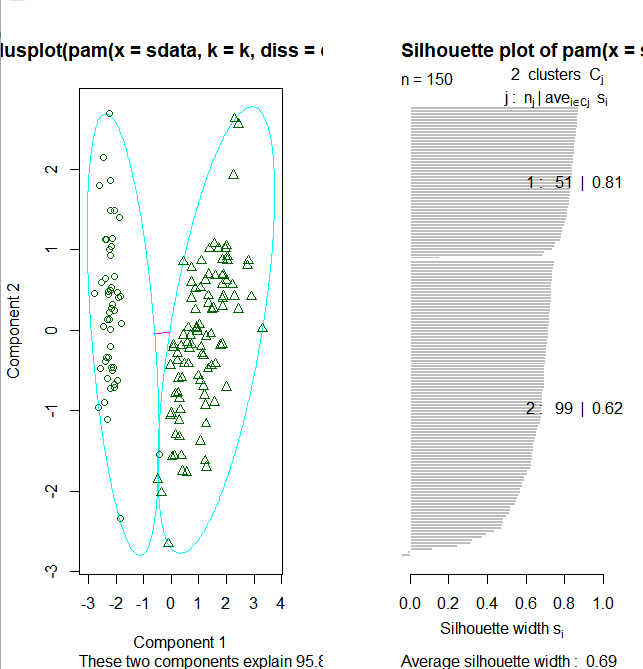
上图右边的图像显示了两个簇的阴影,当si的值较大时(接近1)表明相应的观测点能够正确的划分到相似性较大的簇中,从上面的例子我们可以看出两个簇的si均值分别为0.81和0.62.划分结果很好。
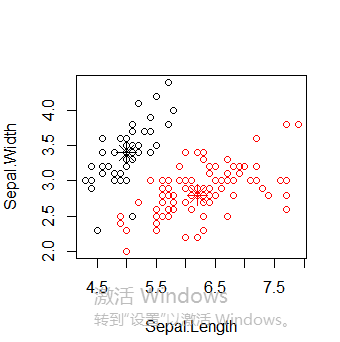
接下来使用函数pam(),并设置k=3.
> library(cluster) > pam.result <- pam(iris2, 3) > table(pam.result$clustering, iris$Species) setosa versicolor virginica 1 50 0 0 2 0 48 14 3 0 2 36
layout(matrix(c(1,2),1,2)) # 每页两张图 plot(pam.result) layout(matrix(1)) #改回每页一张图
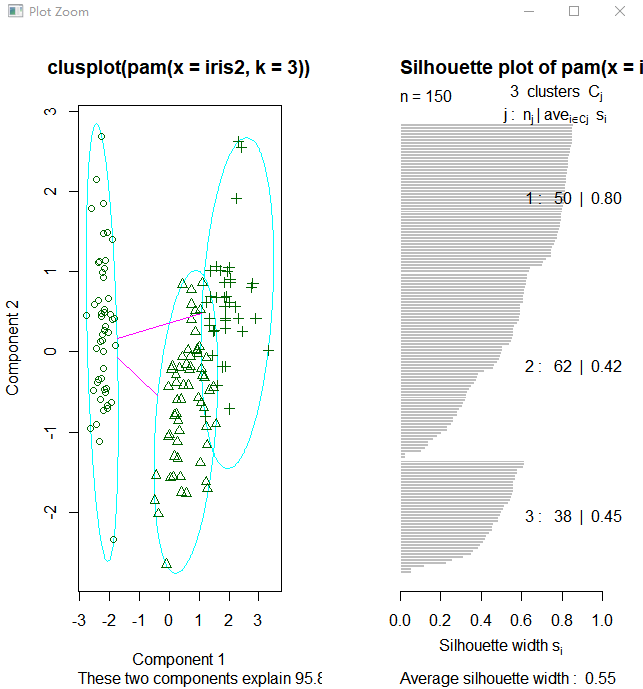
从上面看似乎函数pam()得到的聚类结果更好,因为pam()人工设置识别出三个不同的簇,而pamk()则是利用启发方法识别簇个数。所以很难说谁好谁坏
三、层次聚类
层次聚类是另一种主要的聚类方法,也被称为系统聚类,它具有一些十分必要的特性使得它成为广泛应用的聚类方法。层次聚类的主要思想是:首先每个观测点自成一类;然后,度量所有的观测点彼此间的紧密程度,将其中最亲密的观测点聚成一个小类,形成n-1类,接下来再次度量剩余的观测点和小类间的亲密程度,并用当前最亲密的观测点或小类聚成一类;重复上述过程,不断将观测点与小类聚成大类。层次聚类也可以理解为生成一系列嵌套的聚类树来完成聚类。单点聚类处在树的最底层,在树的顶层有一个根节点聚类。根节点聚类覆盖了全部的所有数据点。首先,我们随机抽取一个40条记录的样本,与之前相同,从数据样本中剔除species属性。
idx <- sample(1:dim(iris)[1], 40) irisSample <- iris[idx,] irisSample$Species <- NULL hc <- hclust(dist(irisSample), method="ave") plot(hc, hang = -1, labels=iris$Species[idx]) rect.hclust(hc, k=3) groups <- cutree(hc, k=3)
数据预处理:随机抽取一个40条记录的样本,与之前相同,从数据样本中剔除species属性
idx <- sample(1:dim(iris)[1], 40) irisSample <- iris[idx,] irisSample$Species <- NULL hc <- hclust(dist(irisSample), method="ave") plot(hc, hang = -1, labels=iris$Species[idx])
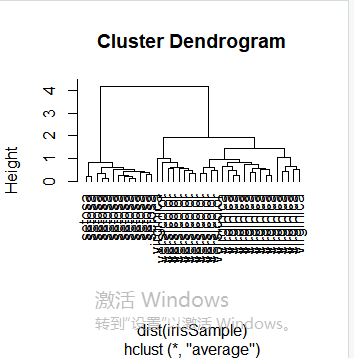
使修剪树使之分为3类
rect.hclust(hc, k=3)
groups <- cutree(hc, k=3)

修剪树使之分为3类

 浙公网安备 33010602011771号
浙公网安备 33010602011771号