[综述阅读] Recent advances in the mechanisms of NLRP3 inflammasome activation and its inhibitors
Recent advances in the mechanisms of NLRP3 inflammasome activation and its inhibitors
本篇综述题为NLRP3炎性小体激活机制及其抑制剂研究进展,来自浙农林的两位教授,浙大的两位教授,以及南京仁信化工的研究员于 2019 年发布在 nature portfolio 子刊 cell death & disease
已被提出的 NLRP3 激活机理
离子流
离子流机理包括 \(K^+\) 流出、\(Ca^{2+}\) 信号传导、\(Na^+\) 流入和氯化物流出,已经被确定为NLRP3炎症体激活的关键事件。相关的信号传递途径可用下图表示:
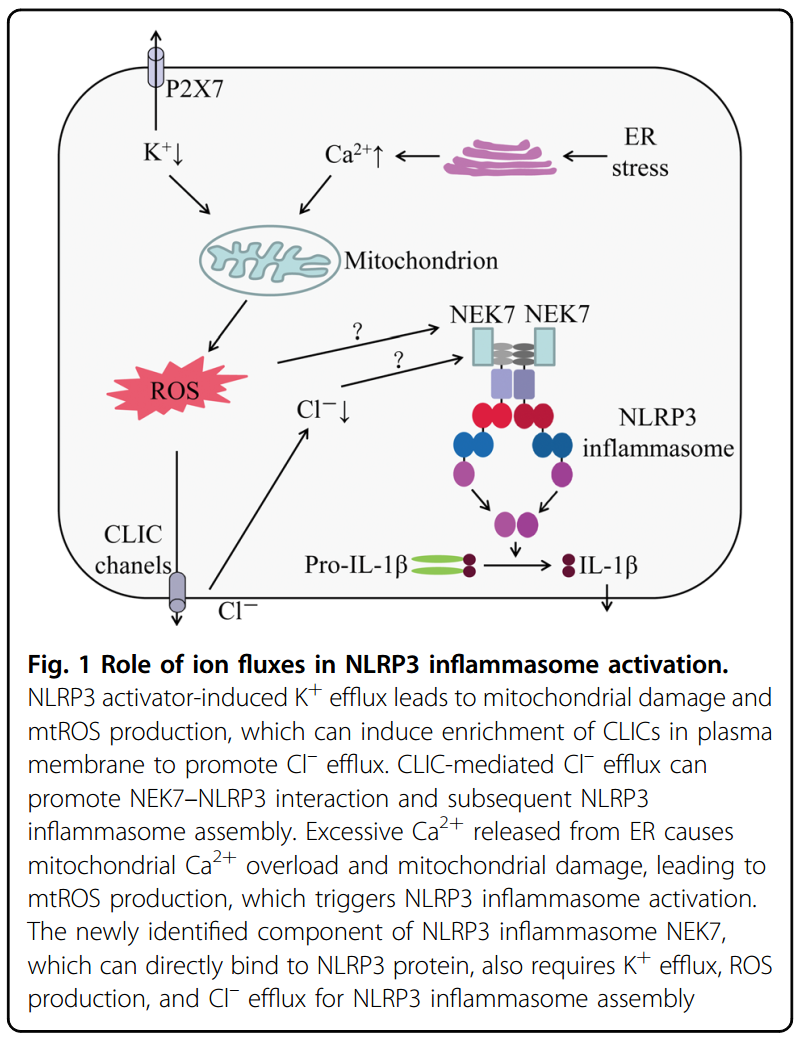
\(K^+\) 流出
由于NLRP3炎性小体激活剂的多样性,NLRP3不太可能直接与刺激物相互作用。细胞内 \(K^+\) 浓度降低首先被确定为 NLRP3 炎症体激活的常见触发因素
事实上,已知许多 NLRP3 炎性小体激活剂可诱导 \(K^+\) 流出,细胞内 \(K^+\) 减少是 NLRP3 炎症小体激活的上游事件
\(K^+\) 外排是 NLRP3 炎性体激活中的一个重要事件,但不是一个特定事件。NLRP3 炎性小体NEK7的一个新发现的成分可以直接结合NLRP3蛋白,也需要 \(K^+\) 外排才能组装 NLRP3 炎症小体。但 \(K^+\) 外排并不特定于 NLRP3 炎性体激活。最近的研究表明,包括咪喹莫特和 CL097 在内的几种小分子可以诱导 ROS 的产生,促进 NLRP3 炎性体的激活,而与 \(K^+\) 外排无关。此外,炭疽杆菌的炭疽致死毒素诱导的 NLRP1b 炎性体激活还需要细胞内 \(K^+\) 降低以激活炎性体和分泌 IL-1β19。
\(Ca^{2+}\) 信号传导
据报道, \(Ca^{2+}\) 信号传导是 NLRP3 炎性体激活所必需的。抑制 \(Ca^{2+}\) 动员会降低NLRP3炎症小体的激活,但对 NLRC4 和 AIM2 炎症小体的活化没有影响
肌醇1,4,5-三磷酸(IP3)是磷脂酶C(PLC)介导的磷脂酰肌醇4,5-二磷酸水解的产物,可以与内质网上的受体 IP3R 相互作用,促进 \(Ca^{2+}\) 动员和 NLRP3 炎症小体激活
然而, \(Ca^{2+}\) 动员如何诱导 NLRP3 炎性小体激活尚不清楚。据推测,内质网过度释放 \(Ca^{2+}\) 会导致线粒体 \(Ca^{2+}\) 超载和线粒体损伤,从而导致线粒体 ROS 的产生,线粒体 ROS 是激活 NLRP3 炎性小体的中枢触发器
相反,其他研究表明, \(Ca^{2+}\) 信号传导对于 NLRP3 炎性小体的激活是不必要的。BAPTA 是一种强 \(Ca^{2+}\) 螯合剂和细胞质 \(Ca^{2+}\) 缓冲剂,可以抑制NLRP3炎性小体激活和 IL-1β 的加工,而与其作为 \(Ca^{2+}\) 螯合物的功能无关
最近的一项研究进一步表明,2-氨基乙氧基二苯硼酸酯(2 APB)是一种具有多个靶点的细胞渗透性 \(Ca^{2+}\) 稳态小分子抑制剂,能够抑制 NLRP3 炎性小体的激活,而与它作为 IP3R 抑制剂的功能无关
储存 \(Ca^{2+}\) 的主要细胞器是内质网和高尔基体,它们在静息状态下维持 \(Ca^{2+}\) 浓度方面起着重要作用,也是特定刺激物释放 \(Ca^{2+}\) 的来源。然而,溶酶体也通过充当响应生理第二信使的 \(Ca^{2+}\) 储存库参与 \(Ca^{2+}\) 稳态的调节,并可以提供与 ER \(Ca^{2+}\) 储存器官的双向通信。因此,需要进一步的研究来阐明 NLRP3 炎性小体激活中 \(Ca^{2+}\) 信号传导的机制
\(Na^+\) 流入
\(Na^+\) 内流是参与NLRP3炎性体激活的另一个重要离子。Schorn 等人报道,MSU 晶体刺激增加了钠离子负荷和细胞肿胀,然后细胞通过水流入被动平衡,这将 \(K^+\) 降低到阈值以下,导致NLRP3炎性体激活
另一项研究表明,细胞外 \(Na^+\) 的流入和细胞内 \(K^+\) 的流出是 NLRP3 炎性体激活对刺激反应的必要条件,\(Na^+\) 内流诱导的 NLRP3 炎性小体激活似乎依赖于 \(K^+\) 外排
然而,\(Na^+\) 离子载体莫能菌素诱导的 \(Na^+\) 内流不会导致 NLRP3 炎性小体激活15。因此,\(Na^+\) 的流入可能不是 NLRP3 炎性体激活的绝对要求
氯化物流出
Verhoef 等人首次报道了氯化物外排在 NLRP3 炎性体激活中的作用。他们表明,降低细胞外 \(Cl^-\) 的浓度可以诱导细胞内 \(Cl^-\) 流出,促进 ATP 诱导的胱天蛋白酶-1激活和 IL-1β 的产生。从那时起,研究表明,氯通道抑制剂,包括氟芬那酸、IAA94、DIDS 和 NPPB,可以抑制 NLRP3,但不能抑制 AIM2 或 NLRC4 炎性体的激活
容量调节性阴离子通道(VRAC)最初被报道为调节NLRP3炎性小体激活的关键阴离子通道。最令人信服的证据来自非甾体抗炎药通过VRAC抑制氯化物流出来预防 NLRP3 炎性体激活的观察。Tang 等人进一步证明,另一种阴离子通道,即氯离子细胞内通道(CLIC),可能起到 VRAC 激活剂的作用
发现 CLIC 依赖的氯化物外流是钾外流-线粒体 ROS 轴的下游事件,CLIC 介导的氯化物外流可以促进 NEK7-NLRP3 相互作用和随后的 ASC 寡聚化,但细胞内氯离子外流如何调节 NEK7-NLRP3 相互作用的具体机制尚不清楚。此外,VRAC 和 CLIC 之间的关系需要进一步确认
活性氧(ROS)的产生
ROS 的产生,特别是来自线粒体的 ROS 的产生是 NLRP3 炎性小体激活的最早发现的触发因素之一。
多项研究表明,大多数 NLRP3 炎性体激动剂可以诱导不同类型细胞中线粒体 ROS 的产生。例如,HFD(高脂肪饮食)引起的脂肪酸可以以 AMPK-自噬-ROS 依赖的方式激活 NLRP3 炎性小体。阻止 ROS 产生的化学抑制剂消除了许多刺激诱导的 NLRP3 炎性体激活。此外,导致细胞死亡和线粒体功能障碍的多种激动剂会增加线粒体 DNA 的氧化,从而激活 NLRP3 炎性小体
然而,NLRP3 炎性小体感知 ROS 产生的机制尚不清楚。有人认为,是NEK7 而不是 NLRP3 本身是 ROS 的传感器。咪喹莫特是一种 TLR7 激动剂,可以诱导 ROS,但不能诱导 \(K^+\) 依赖的 NLRP3 炎性体激活;然而,缺乏 NEK7 的细胞在咪喹莫特刺激下未能产生 IL-1β。其他研究还表明,ROS 在启动步骤发挥作用,因为 ROS 特异性抑制剂可以通过在启动步骤干扰 NLRP3 表达来阻断 NLRP3 炎性体的激活,而直接的 NLRP3 激活则不受影响
相比之下,也有报道称线粒体 ROS 非依赖性 NLRP3 炎性小体激活。Jabaut 等人揭示,血清淀粉样蛋白 A 诱导的线粒体 ROS 依赖性和 ROS 非依赖性机制在NLRP3炎性小体 / IIL-1β 分泌轴中发挥作用。NLRP3 激动剂以ROS依赖的方式促进 NLRP3 与硫氧还蛋白相互作用蛋白(TXNIP)的相互作用,但在没有 TXNIP 的情况下,胱天蛋白酶-1的激活和 IL-1β 的分泌并没有完全受到抑制,这表明存在其他机制。因此,需要进一步的研究来阐明 ROS 在 NLRP3 炎性体激活中的确切作用
溶酶体失稳
淀粉样蛋白 β(Aβ)是阿尔茨海默病中表达的一种致病性错误折叠蛋白,是第一种被鉴定为通过溶酶体失稳激活 NLRP3 炎性小体的物质。我们之前的工作和其他研究小组表明,朊病毒病中另一种错误折叠的蛋白质 PrP 原纤维诱导溶酶体失稳和 NLRP3 炎性体激活
Hornung 等人证明大颗粒激活剂(如二氧化硅和明矾)的低效清除会导致溶酶体破裂和组织蛋白酶 B 释放,从而触发 NLRP3 炎性体激活。在胆固醇晶体诱导的 NLRP3 炎性体激活模型中,与野生型相比,组织蛋白酶 B 或 L 缺乏的小鼠产生的 IL-1β 很少。此外,B 组链球菌和5型腺病毒诱导的 NLRP3 炎性体激活也取决于溶酶体渗漏
溶酶体失稳似乎不仅参与激活步骤(信号2),还参与启动步骤(信号1)。在棕榈酸酯诱导的 NLRP3 炎性体激活中,溶酶体钙信号通过稳定 IL-1βmRNA 调节 pro-IL-1β 的产生(信号1),而溶酶体蛋白酶的组织蛋白酶 B 有助于 NLRP3 炎症体激活(信号2)
最近的一项研究进一步证实了这一结果,该研究表明,多种组织蛋白酶可以促进 pro-IL-1β 的合成和 NLRP3 的激活。然而,组织蛋白酶 B 抑制剂可能通过脱靶效应或靶向组织蛋白酶家族的其他成员来防止NLRP3激活。据报道,CA-074-Me 还抑制了炭疽致死毒素诱导的 NLRP1b 炎性体激活和胱天蛋白酶-1切割。
组织蛋白酶 B 缺乏的骨髓源单核细胞(BMDMs)在接受血偶素晶体治疗后,胱天蛋白酶-1切割和 IL-1β 分泌没有差异
Muñoz-Planillo 等人报告称,颗粒物的内化通过吞噬作用导致溶酶体膜损伤,这种损伤可以通过打开一个或多个可渗透 \(K^+\) 的膜孔,引发 \(K^+\) 流出引起的NLRP3炎性体激活。有趣的是,他们还发现LPS启动可能会增强颗粒激活剂(包括LL-OMe、Al(OH)3、SiO2 和 CPPD 晶体)引起的 \(K^+\) 外流
因此,颗粒激活剂诱导溶酶体失稳与 \(K^+\) 外流的确切机制需要完善并确定
NLRP3的翻译后修饰(PTMS)
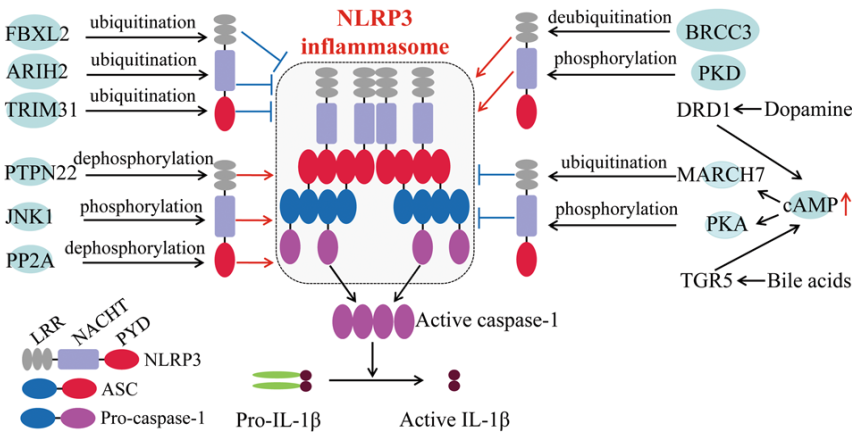
最近的研究表明,NLRP3 的翻译后修饰,包括磷酸化和泛素化,在 NLRP3 炎性小体激活中起着关键作用。
泛素化
Py 等人使用去泛素化的小分子抑制剂 G5 的研究表明,G5 抑制了不同类型激活剂(包括组织蛋白酶、ROS 或 \(K^+\) 外排依赖性激动剂)诱导的 NLRP3 炎症小体激活,但 G5 对 AIM2 和 NLRC4 炎症小体激活没有影响。通过筛选去泛素化酶(DUB)库,他们认识到 BRCC3(含有 BRCA1-BRCA2 的复合亚基3)去泛素了 NLRP3 的 LRR 结构域,然后进行 NLRP3 激活
另一项研究表明,信号1和信号2通路都会刺激 NLRP3 去泛素化。启动步骤可能会激活靶向 NLRP3 特定结构域的 DUB 酶,而 ATP 信号传导可能会激活另一种 DUB 酶来去泛素化不同的结构域。由 TNF-α 诱导蛋白3(TNFAIP3)基因编码的 NFκB 抑制剂 A20 通过泛素化前 IL-1β 的生理位点 K133 来阻止 IL-1β 的自发分泌
最近的一项研究发现,神经递质多巴胺(DA)是 NLRP3 炎性小体的内源性调节因子。DA 及其受体 DRD1(多巴胺 D1 受体)信号传导可以抑制神经毒素诱导的神经炎症。从机制上讲,DA–DRD1 信号通过第二信使 cAMP(环磷酸腺苷)负调控 NLRP3 炎性小体的激活,cAMP 与 NLRP3 蛋白结合,并通过 E3 泛素连接酶 MARCH7 促进其泛素化和降解
另一个参与 NLRP3 炎性体激活的重要 E3 泛素连接酶是 TRIM31。LPS 启动后,TRIM31 和 NLRP3 的表达被诱导,然后 TRIM31 直接与 NLRP3 结合,促进其 K48 连接的泛素化,导致 NLRP3 蛋白酶体降解和抑制。最近的一项研究表明,作为启动阶段的一部分,Pellino2 可以通过促进 NLRP3 的 K63 连接泛素化来促进 NLRP3-inflammatosome 的激活,而 Pellino2 在启动阶段不参与 TLR 诱导的 NLRP3 和 pro-IL-1β 的上调。
相比之下,仅含 F-box 的蛋白3 FBXO3 在启动过程中参与 LPS 诱导的 NLRP3 蛋白上调。另一种 F-box 蛋白 F-box L2(FBXL2)可以与赖氨酸689残基上的 NLRP3 相互作用并泛素化,以促进蛋白酶体降解。因此,TLR刺激会增加 FBXO3 的表达,FBXO3 泛素化并介导 FBXL2 的降解,从而减少 NLRP3 的降解。
另一种 E3 连接酶 Ariadne 同系物2(ARIH2)被报道为巨噬细胞 NLRP3 炎性小体激活的翻译后负调节因子。ARIH2 可以通过 NLRP3 的 NACHT 结构域泛素化 NLRP3,ARIH2的 RING2 结构域是 NLRP3 泛素化所必需的。ARIH2 的过表达促进 NLRP3 泛素化并抑制 NLRP3 炎性体激活
蛋白质的泛素化可以通过不同的途径通过磷酸化来调节,包括促进 E3 连接酶的识别或调节底物和连接酶的相互作用。胆固醇分解代谢衍生的胆汁酸可以被 TGR5 受体(跨膜G蛋白偶联受体-5)感知,这种相互作用增加了细胞内 cAMP 水平,随后激活了 PKA(蛋白激酶A)。PKA 催化亚基与 NLRP3 全长结合,并在 Ser291 上磷酸化其 NACHT 结构域。Ser291 的磷酸化反过来促进 K48 和 K63 连接的多泛素化和 NLRP3 降解,从而抑制 NLRP3 炎性体激活。几乎同时,Mortimer 等人证实了人类 NLRP3 上相同的 PKA 磷酸化位点Ser295(对应于小鼠中的NLRP3 Ser291)
泛素化
然而,同一残基的磷酸化可能会引发相反的效果。例如,Zhang 等人证明,NLRP3 炎性小体刺激促进线粒体相关膜(MAM)定位到相邻的高尔基体膜和二酰基甘油(DAG)积累。高尔基体上的DAG积累激活蛋白激酶D(PKD),随后磷酸化 NLRP3,导致完全成熟的炎性小体组装。这强烈表明,NLRP3 磷酸化的后果很可能取决于这种修饰何时何地发生
磷酸化也可能发生在启动步骤,这是 NLRP3 转录所必需的。然而,最近的研究表明,NLRP3 炎性小体的激活可以独立于转录而触发,这表明该过程中存在未知的关键调控步骤。Song 等人证明,在启动步骤中,NLRP3通过 JNK1 介导的 S194 处的NLRP3磷酸化而磷酸化,S194 是去泛素化上游 NLRP3 炎性体激活的关键调节因子
除了丝氨酸磷酸化外,NLRP3 的酪氨酸磷酸化也被报道参与调节 NLRP3 炎性体的激活。PTPN22(蛋白酪氨酸磷酸酶非受体22)与 NLRP3 相互作用,并在 Tyr861 处对其进行去磷酸化,从而有效激活 NLRP3 炎性体和分泌IL-1β,PTPT22 不影响 AIM2 和 NLRC4 炎性体的激活
此外,PYD 结构域的磷酸化可能会中断 NLRP3 和 ASC 之间的相互作用。Stutz 等人证明,PP2A(磷酸酶2A)使位于PYD-PYD相互作用界面的 NLRP3 的 Ser5 去磷酸化,该位点的磷酸化残基抑制 NLRP3-ASC 以及 NLRP3-PYD-PYD 相互作用
因此,NLRP3 的磷酸化在调节 NLRP3 炎性体激活中起着关键作用,但磷酸化的差异可能取决于启动时间、刺激和细胞类型。研究其他翻译后修饰,如乙酰化、甲基化和琥珀酰化,在调节 NLRP3 炎性体激活中的作用也将是很有意义的
非典型炎性体激活
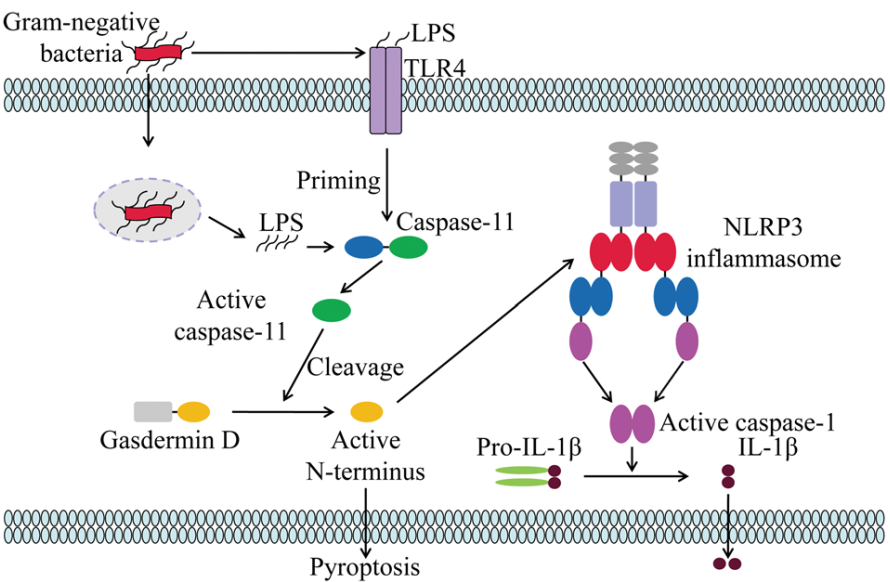
人和小鼠中胱天蛋白酶 -4/5 和胱天蛋白酶 -11 的激活途径称为非正常炎症体激活。规范和非规范炎症小体激活模式最终都会导致细胞溶解和促炎细胞因子的释放,但它们的机制存在显著差异。与典型的 NLRP3 炎症小体激活不同,非典型炎症小体激活是由小鼠的 caspase-11 和人类的 caspase-4/5 触发的
然而,由于静息细胞中 caspase-11 的低表达水平,诱导 caspase-11 转录的启动信号对于非非典型炎性体激活也是必不可少的。在人类中,胱天蛋白酶-4在许多非单核细胞和单核细胞中组成型表达;因此,细胞质 LPS 可以在没有启动步骤的情况下激活非正常炎症小体
非典型炎性体可以感应到许多革兰氏阴性菌,但不能感应到革兰氏阳性菌,这表明革兰氏阴性菌的外膜 LPS 可能是非非典型炎症体的关键激活剂。进一步的研究表明,LPS的保守区脂质A是非正常炎性体激活的原因
细胞内 LPS 或脂质A直接被胱天蛋白酶4/5/11的 CARD 结构域识别,导致其寡聚化,随后在N端和C端结构域之间的接头内,由活性胱天蛋白酶切割成孔蛋白 gasdermin D。GSDMD 释放的N-末端结构域靶向质膜,形成内径为10-14nm的膜孔,这有助于钾外流、焦亡和随后的 NLRP3 炎性体激活
因此,胱天蛋白酶4/5/11不会切割白细胞介素,只会导致焦亡,随后钾外排诱导的 NLRP3 炎性小体激活是胱天蛋白酶-1激活和 IL-1β 分泌的原因。这一证据表明了规范和非规范炎症小体之间的相互作用
替代性炎性体激活
众所周知,NLRP3 炎性小体激活通过两步机制触发胱天蛋白酶-1激活和 IL-1β 成熟。然而,令人信服的证据表明,单独使用 LPS 足以诱导人单核细胞中依赖胱天蛋白酶-1的 IL-1β 成熟和产生
Gaidt 等人首次发现了一种新型的炎性小体激活,并将其命名为选择性炎性小体活化,该活化由TLR4信号通路诱导,不涉及其他第二激活因子
在替代激活中,NLRP3 是主要因素,其激活需要 TRIF(含衔接蛋白TIR结构域的衔接分子1)和随后胱天蛋白酶-8的切割
替代性和经典性炎性体激活(规范性和非规范性)之间的主要区别包括对K+流出的非依赖性、没有焦小体形成和焦亡。此外,另一种炎性体激活是物种特异性的,因为它仅限于人类和猪的单核细胞,但在小鼠细胞中没有观察到
信号通路依赖于TLR4-TRIF-RIPK1-FADD-CASP8来促进 NLRP3 炎性体的激活。然而,该信号轴仅限于另一种炎性小体,在经典的 NLRP3 炎性小体激活中没有作用, caspase-8 在选择性炎症小体激活期间激活 NLRP3 的确切机制尚不清楚
据推测,胱天蛋白酶-8介导的未知中间蛋白的激活是替代性炎性体激活所必需的。因此,需要进一步的研究来确定胱天蛋白酶-8和 NLRP3 在选择性炎性体激活过程中缺失的联系。
线粒体DNA(mtDNA)的新合成
如上所述,鉴于NLRP3炎性小体激动剂的多样性,NLRP3似乎可能感知到由细胞内过程诱导的共同触发途径。Zhenyu Zhong 等人的最新研究表明氧化mtDNA(oxmtDNA)的产生可能是NLRP3炎性体激活的“最终”配体
众所周知,线粒体损伤会导致ox-mtDNA的产生,是NLRP3激活的关键事件,但线粒体损伤不能在没有启动的情况下触发NLRP3的激活。Zhong等人证明了线粒体如何在NLRP3触发过程中将启动和激活阶段联系起来
LPS启动可以增加线粒体中的 mtDNA 合成,这取决于 TLR 接头 MYD88(在早期时间点)、TRIF(在后期时间点)和两个接头下游的 IRF1
新合成的 mtDNA 负责产生 ox-mtDNA,在 NLRP3 刺激下与 ASC 共定位。他们进一步确定线粒体脱氧核糖核苷酸激酶 UMP-CMPK2(CMPK2)是 IRF1 的下游靶标,CMPK2 是控制 LPS 诱导的 mtDNA 合成的 dNTP 前体供应的限速酶
因此,作者确定了 TLR-MyD88/TRIF-IRF1-CMPK2 轴在 NLRP3 炎性体激活中的关键作用,并提供了可用于治疗 NLRP3 依赖性疾病的新策略。

 浙公网安备 33010602011771号
浙公网安备 33010602011771号